
Abstract
Species boundaries and geographic distribution of corticioid fungi (resupinate Basidiomycota) are often poorly known. Our recent study on Subulicystidium showed that species diversity in this genus is at least twice as high as previously recognized. This re-estimation of the species diversity was based on a study of only a part of the genus. The present study sheds light on molecular and morphological diversity of three more species. We generated 27 ITS and 24 28S nuclear ribosomal DNA sequences from 49 specimens labelled as Subulicystidium cochleum, S. longisporum and S. perlongisporum and collected in distant geographic localities. We assessed pairwise dissimilarities and phylogenetic relationships of DNA sequences with Bayesian and maximum likelihood methods. We correlated phylogenetic information with morphological data on spores and cystidia. We found that the three species are not closely related, despite their similarity in spore shape and size. In one of the species, S. perlongisporum, we detected the presence of two sympatric lineages. These lineages are not morphologically distinct, except for a small difference in the mean length of cystidia. Our study provides a further example of transoceanic species distribution in Agaricomycetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Species boundaries and geographic distribution of corticioid fungi (resupinate Basidiomycota) are often poorly known. Fungi from the genus Subulicystidium Parmasto 1968 (Hydnodontaceae, Trechisporales, Agaricomycetes, Basidiomycota) occur on moderately or strongly decayed wood and are common in many forest ecosystems, especially tropical ones. For many decades, Subulicystidium has challenged both morphology- and DNA-based mycology. The presence of numerous transitional forms as to basidiospore size and shape hindered species delimitation (Oberwinkler 1977; Liberta 1980; Duhem and Michel 2001). Recently, Ordynets et al. (2018) described 11 new species based on analyses of ribosomal DNA and morphometry of spores and cystidia. The genus now includes 22 accepted species (Index Fungorum 2019).
In Subulicystidium, morphological species identification through a key traditionally begins with defining whether the spores’ length-to-width ratio (Q) is lower or higher than four. This criterion is stable and allows one to assign each specimen unambiguously to one of the morphogroup (Boidin and Gilles 1988; Duhem and Michel 2001; Gorjón et al. 2012). For easier communication, Ordynets et al. (2018) labelled these morphogroups as “short-spored” (Q < 4) and “long-spored” (Q > 4). In that study, the authors focused exclusively on short-spored taxa. Liu et al. (2019) provided an insight into the diversity of the long-spored morphogroup by describing two new species from East Asia (Subulicystidium acerosum S.H. He & S.L. Liu and S. tropicum S.H. He & S.L. Liu).
The present study focuses on additional long-spored species, viz. Subulicystidium cochleum Punugu, S. longisporum (Pat.) Parmasto and S. perlongisporum Boidin & Gilles. S. longisporum is the oldest described species and also the type of the genus. S. cochleum was claimed to differ from S. longisporum by the presence of needle-like crystals on cystidia and by more strongly curved spores (Punugu et al. 1980). S. perlongisporum was described by Boidin and Gilles (1988) and differs from S. longisporum by more elongated spores. The holotype of S. longisporum is from Tunisia (Patouillard 1894) and the type of S. perlongisporum is from La Réunion Island. Both species have been recorded from several continents as well as from islands in the Caribbean, Indian Ocean and the Pacific. The holotype of S. cochleum is from Saint Lucia Island in the Caribbean and outside that region was only reported from Hawaii (Martini 2020). Almost all records of these species are based on the observation of fruiting bodies and few DNA sequences are available.
In this study, we use molecular data to answer (1) whether S. cochleum, S. longisporum and S. perlongisporum can be considered as distinct and monophyletic species and (2) whether the similarity in spore shape and size is reflected by the phylogenetic position. For that, we explore the variation and relationship of DNA sequences of the nuclear ribosomal ITS and 28S region from geographically scattered herbarium specimens. We also correlate molecular data with the measurements of basidiospores and cystidia.
Materials and methods
Microscopy
In this study, we examined 49 herbarium specimens: 37 identified as Subulicystidium perlongisporum, seven as S. cochleum and five as S. longisporum. They are preserved in the following herbaria: ARAN, FR, GB, KAS, KR, MSK, LE, LY, MG, O, PC, SP, TU and TUB (acronyms, follow http://sweetgum.nybg.org/science/ih). Detailed information on the studied collections is provided in Supplementary file 1.
Sections from dried herbarium specimens were examined in 3% aqueous solution of potassium hydroxide (KOH) mixed with 1% aqueous solution of Phloxine at × 1000 magnification, using a Leica DM500 light microscope. Images were captured with a built-in ICC 50 HD Camera using Leica Application Suite EZ software versions 3.2.1 and 3.4.2 (Leica Microsystems GmbH, Wetzlar, Germany). Measurements were done with the software “Makroaufmaßprogramm” from Jens Rüdigs (Rüdig 2019, https://ruedig.de/tmp/messprogramm.htm) and processed with the software “Smaff” v. 3.2 (Wilk 2012).
Where possible at least 30 basidiospores per specimen were measured for all 36 sequenced specimens (28 specimens of S. perlongisporum, 5 of S. longisporum and 3 of S. cochleum). For each specimen, an automated search for size outliers, and their exclusion was performed as described in Ordynets et al. (2018). This resulted in 1203 measurements after excluding outliers (see Supplementary file 2). Spore sizes were divided into a 90% main range, and 5% lowest and highest values as described in Ordynets and Denecke (2018), by means of the package “dplyr” (Wickham et al. 2019) in R v. 3.5.3 (R Core Team 2019).
During the measurements of cystidia, the protruding bow-tie crystals were included in the cystidium diameter. In most cases, between 9 and 28 cystidia per specimen were measured (in two cases nine and in one case 38). In total 645 cystidia were measured from 36 sequenced specimens (see Supplementary file 2). These measurements were not checked for outliers and were directly summarized on specimen level with the same method as used for the spores (Ordynets and Denecke 2018). To compare the mean size and shape of spores and cystidia between phylogenetically defined groups, the unpaired two-sample Wilcoxon test was used as implemented in the function “wilcox.test”, with default settings, from the R “stats” package (R Core Team 2019).
DNA extraction, amplification and sequencing
Sequences of two nuclear ribosomal DNA regions were considered in our study: internal transcribed spacer (nc ITS rDNA) and ribosomal large subunit-coding DNA (nc 28S rDNA). Total DNA was extracted from dried herbarium specimens. For this, pieces of fungal fruiting bodies totalling about 20 mm2 were placed into 2-ml tubes containing two small and two large sterile metallic beads. The tube content was ground in the mixer mill for 1 min at 30 Hz. Then, tubes were centrifuged at 16,060g for 30 s. The following methods of total DNA isolation were used:
-
(1)
Most of the specimens were processed with the E.Z.N.A.® Fungal DNA Mini Kit from Omega Bio-tek, Inc. (Norcross, GA, USA). We used the short version of the protocol from the manufacturer’s manual with few modifications. In particular, after adding FG1 buffer, we added to each tube 10 μl of proteinase K (20 mg/ml, Ambion, Thermo Fischer Scientific, Waltham, MA, USA) and used neither RNase A or β-mercaptoethanol. We also used a fixed volume of 300 μl for FG3 buffer and 600 μl for 100% ethanol. We added sterile deionized water instead of elution buffer at the two final elution steps. We performed all the centrifuging steps at 9500g. We used the DNA gained with this protocol in undiluted form for PCR.
-
(2)
For Reunionese specimens collected in 2013 and 2015 (herbaria KAS, FR), the protocol of Izumitsu et al. (2012) was used (see details in Ordynets et al. 2018).
-
(3)
For collections from Russia (herbarium LE), total DNA extraction followed the manufacturer’s protocol of the NucleoSpin Plant II kit (Macherey–Nagel GmbH and Co. KG, Düren, Germany).
-
(4)
In specimens from Sicily and Estonia (herbarium TU), the total DNA was extracted in a lysis buffer (0.8 M Tris-HCl, 0.2 M (NH4)2SO4, 0.2% w/v Tween-20) (Soil BioDyne, Tartu, Estonia) using a proteinase K method (100 μl lysis buffer and 2.5 μl proteinase K; incubation at 56 °C for 24 h and at 98 °C for 15 min).
Primer pairs used to amplify the complete ITS region were ITS1f/ITS4, ITS1/ITS4, ITS1f/ITS4B and ITS5/ITS4 (White et al. 1990; Gardes and Bruns 1993). If the amplification of the complete ITS region failed, it was performed for shorter ITS portions with primer pairs ITS1/ITS2 and ITS3/ITS4. Each of these two fragments represents nearly half of ITS and they were later assembled to a single complete ITS sequence. The D1–D2 domains at the 5′ end of the 28S region were amplified with primer pair LR0R/LR5 (Hopple and Vilgalys 1999) or alternatively with NL1/NL4 (O’Donnell 1992). Finally, for some Sicilian collections, primer pair ITS1OF/LB-W was used to recover the full ITS and partial 28S region with a total length of ca. 1000 nucleotides (Tedersoo et al. 2008).
The PCR after extraction methods 1 and 2 were performed on 25 μl solution containing 2.5 μl of extracted DNA and 22.5 μl master mix. One master mix portion contained 15.1 μl of double-distilled H2O, 5 μl of 5× MangoTaq™ Colored Reaction Buffer, 1 μl dNTPs (5 mM), 1 μl MgCl2 (50 mM), 0.1 μl MangoTaq DNA polymerase 5 units/μl (all components above from Bioline GmbH, Luckenwalde, Germany), 1 μl of bovine serum albumin (20 μg/μl) and 0.4 μl of each forward and reverse primers (25 pmol) from Thermo Fisher Scientific (Waltham, MA, USA). PCR with primer pairs ITS1F/ITS4 and ITS3/ITS4 was set as initial denaturation at 94 °C for 3 min followed by 29 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 45 s and extension at 72 °C for 60 s; final elongation was done at 72 °C for 7 min. PCR with primer pair LR0R/LR5 differed in having the annealing temperature as 58 °C and for primer pairs NL1/NL4 and ITS1/ITS2 as 60 °C. PCR products were checked on 1% agarose gel stained with GelRed fluorescence dye (Biotium, Hayward, CA, USA). PCR products were cleaned with QIAquick PCR Purification Kit according to the manufacturer’s instructions (QIAGEN GmbH, Hilden, Germany). Sanger sequencing of purified products was performed by the company Eurofins Genomics Germany GmbH (Ebersberg, Germany) and in the facilities of the Senckenberg Research Institute and Natural History Museum (Frankfurt am Main, Germany). The primers used for sequencing were identical to those used for amplification.
The PCR after extraction method 3 was performed on 20 μl solution containing 2 μl of extracted DNA, 7.6 μl of distilled H2O and 10 μl of iQ Supermix (Bio-Rad Laboratories, USA). The purification of PCR products was made with the GeneJET PCR Purification Kit (Thermo Fisher Scientific, Lithuania). Sequencing was performed in house on ABI 3130 Genetic Analyzer (Applied Biosystems, CA, USA).
The PCR after extraction method 4 was performed in 25 μl containing 0.5 μl of each primer, 5 μl FirePol Mastermix (Solis BioDyne, Tartu, Estonia), 1 μl of 10 times diluted DNA template and sterilized distilled water. PCR conditions for the amplification of ITS region were set as initial denaturation at 95 °C for 15 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s and extension 72 °C for 60 s; final elongation was done at 72 °C for 7 min. PCR products were cleaned using Exo-SAP enzymes (GE Healthcare, Freiburg, Germany) using incubation at 37 °C for 45 min and at 85 °C for 15 min. PCR products were sequenced by Macrogen Inc. (Amsterdam, the Netherlands) with the same primers as used for PCR.
DNA sequence–based analyses
Raw sequence data were processed with Geneious v. 5.6.7 (http://www.geneious.com, Kearse et al. 2012) and Sequencher v. 5.1 (Gene Codes Corporation, Ann Arbor, USA). For various sequence format conversion and alignment viewing, Mesquite v. 3.6 (build 917) (Maddison and Maddison 2018) and AliView v. 1.19 (Larsson 2014) were used. For phylogenetic data import and processing within R, the following packages were used: “ape” (Paradis et al. 2004), “forcats” (Wickham 2019), “gdata” (Warnes et al. 2017), “geiger” (Harmon et al. 2008), “Hmisc” (Harrell Jr et al. 2019, and with contributions from Charles Dupont, many others 2019), “pals” (Wright 2018) and “treeio” (Yu 2019).
In this study, the following numbers of new nc ITS rDNA sequences were generated: 21 sequences for S. perlongisporum, three for S. cochleum and three for S. longisporum. For the nc 28S rDNA, the numbers of sequences generated for these same species were 19, 3 and 2, respectively. The newly generated DNA sequences were submitted to GenBank (Benson et al. 2018). Additional six ITS and four 28S sequences of S. perlongisporum and two ITS and three 28S sequences of S. longisporum available in GenBank (e.g. Volobuev 2016) and UNITE database (Nilsson et al. 2018) were used in our analyses after the corresponding herbarium specimens were examined (Table 1).
To relate the DNA sequences of S. perlongisporum, S. cochleum and S. longisporum to the rest of the genus Subulicystidium, we combined our data for these three species with the datasets of 57 ITS and 55 28S DNA sequences of other Subulicystidium species (Table 2). We used ITS and 28S DNA sequences from the holotype of S. oberwinkleri (KAS:L 1860) as an outgroup in all genus-level phylogenetic analyses.
Sequences from each locus, 92 of ITS region and 86 of 28S region, were aligned in MAFFT v. 7 online (Katoh et al. 2017), with L-INS-i algorithm and other settings as default.
The small fragments of 18S rDNA and 28S rDNA were automatically trimmed from the target ITS region with the ITSx software (Bengtsson-Palme et al. 2013) implemented in the PlutoF workbench (Abarenkov et al. 2010). The final ITS alignment had 659 nucleotide positions. The 28S alignment was trimmed manually to produce sequences of the same length and with fewer (if any) gaps at both ends, leaving 911 positions in the final version. ITS and 28S alignments were concatenated with the SequenceMatrix v. 1.8 (Vaidya et al. 2011) to produce a matrix with 100 rows and 1570 columns.
We performed phylogenetic reconstruction for Subulicystidium from concatenated ITS+28S alignment using Bayesian and maximum likelihood analyses. For Bayesian inference of phylogeny, GTR+G+I evolutionary model was used in MrBayes 3.2.3 (Ronquist et al. 2012) run on CIPRES Science Gateway v. 3.3 (Miller et al. 2010; http://www.phylo.org). Two independent MCMC processes, each in 4 chains, were run. Ten million trees were generated; the sample frequency was set to 1000 and burn-in fraction to 0.25. For 15,002 sampled trees (burn-in fraction excluded), a majority-rule consensus tree was computed with branch supports representing the relative frequencies of bipartitions (posterior probabilities, PP). This analysis was finished with the standard deviation of split frequencies of 0.0088 (equals average) and was characterized for branch and node parameters by the potential scale reduction factor between 0.999 and 1.003. The median of the log-likelihood for the sampled trees from two MCMC runs equalled − 11,344.25 and the pooled effective sample size was 4924.1347. The plot of the generation versus the log-likelihood values of the sampled trees was inspected with Tracer v. 1.6 (Rambaut 2014) to confirm the stationarity of the tree samplings.
Maximum likelihood analysis of concatenated ITS+28S alignment was performed with PhyML v. 3.0 (Guindon et al. 2010) after automated model selection with SMS v. 1.8.1, both run online (Lemoine et al. 2019, https://ngphylogeny.fr). Evolutionary model comparison was performed under Bayesian information criterion and resulted in selecting TN93+G+I as the best-fitting model. BioNJ was used as a starting tree. The algorithm to explore the space of tree topologies was subtree pruning and regrafting (SPR). For branch support estimation, an approximate likelihood ratio test was requested that relies on a nonparametric, Shimodaira-Hasegawa-like procedure (SH-like aLRT, Guindon et al. 2010).
To visualize results for the genus-level concatenated ITS+28S dataset, the consensus tree from Bayesian analysis was plotted with both posterior probabilities from Bayesian analysis and SH-like aLRT support values from PhyML. For this, we used R script of Ordynets which is based on the R code from Crane (2013).
In the most abundant species, S. perlongisporum, we analyzed also the intraspecific genetic diversity. We produced a species-level ITS alignment with 27 sequences that after trimming with ITSx was 554 positions long. We calculated pairwise genetic distances between these ITS sequences using “pairDistPlot” function of “adegenet” R package (Jombart 2008) setting the arguments as follows: model = “raw”, pairwise.deletion = TRUE (i.e. with deleting the sites with missing data in a pairwise way). Phylogenetic inference for this dataset was performed in PhyML v. 3.0 online with settings identical to those in the analysis of concatenated genus-level dataset but with HKY85+G as the best model. The result was visualized with the R packages “ggtree” (Yu et al. 2017), “ggstance” (Henry et al. 2019) and “ggplot2” (Wickham 2009).
The multiple sequence alignments, details of phylogenetic analyses and trees were deposited in TreeBASE (Piel et al. 2009) under submission ID S24881. The R code for the DNA-based analyses and visualizations is available on GitHub (https://github.com/ordynets/subLongSpored).
Results
Phylogenetic analyses
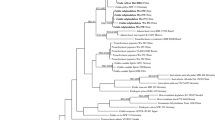
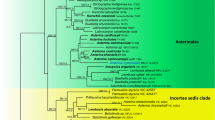
The genus-level phylogenetic analyses based on the ITS+28S dataset showed that each of the species Subulicystidium cochleum, S. longisporum and S. perlongisporum is monophyletic (Fig. 1). The three species were not closely related to each other. S. cochleum (branch support PP = 1, aLRT = 0.92) was placed as a sister species to S. acerosum on a highly supported branch (PP = 1, aLRT = 1). S. longisporum (PP = 1, aLRT = 0.95) was nested in the clade dominated by numerous sequences of S. meridense and S. brachysporum (PP = 0.94, aLRT = 0.87). S. perlongisporum (PP = 1, aLRT = 0.98) was most closely related to S. robustius and S. rarocrystallinum (occupying the next more basal node) and to S. boidinii, S. harpagum, S. parvisporum and S. tropicum and to the specimen of S. obtusisporum from Jamaica (occupying the next highest node).

Phylogenetic relationship of Subulicystidium based on concatenated ITS+28S nc rDNA alignment. 50% majority-rule consensus tree from Bayesian analysis is shown, with posterior probabilities above the branches and supports from approximated likelihood ratio test from the maximum likelihood estimation below the branches, both in the range from 0 to 1. NA means the absence of branch support. Tips of the tree include GenBank/UNITE accession numbers for the ITS region followed by 28S region, voucher specimen and country of collection. Tree tips are coloured according to the species name. Scale bar shows the number of substitutions per site
Concatenated ITS+28S DNA sequences from S. perlongisporum were grouped into two clades of unequal size (Fig. 1). The larger clade included the holotype (LY 11631) and sequences of various geographic origin (Paleotropics, Neotropics and temperate Europe) strongly intermixed. The smaller clade contained two specimens from temperate Europe, two from the Mediterranean region in Europe and one specimen from the Southern Hemisphere (La Réunion).
Within the larger clade of S. perlongisporum, further labelled as clade 1 (aLRT = 1), two subclades on short basal branches were distinguishable (Fig. 2a). Subclade 1.1 (aLRT = 0.8) included DNA sequences merely from the Eastern Hemisphere while subclade 1.2 (aLRT = 0.67) was dominated by DNA sequences from the Western Hemisphere. The sequences from La Réunion and Madagascar were found in both subclades.

Relationship of ITS nrDNA sequences of Subulicystidium perlongisporum (a) and corresponding multiple sequence alignment (b). Maximum likelihood phylogenetic tree includes branch supports from approximated likelihood ratio tests. Scale bar (a): number of substitutions per site, (b) DNA sequence length in bases
Genetic and morphological diversity within S. perlongisporum
Between the ITS rDNA sequences of S. perlongisporum, there were signatures in the multiple sequence alignment that allowed to differentiate clade 2 from clade 1. The most striking feature of clade 2 was the insertion at positions 107–118 (Fig. 2b). Additionally, there was an insertion at positions 381–385 in sequences that formed subclade 1.1 in the ITS tree (Fig. 2a).
Pairwise genetic distances were lowest between members of clade 1 (maximum 0.037, median 0.021; Fig. 3) and slightly higher between members of clade 2 (maximum 0.052, median 0.042). The distances between the members of clades 1 and 2 were always distinctly higher (maximum 0.122, median 0.109).

Genetic distances within and between phylogenetic clades 1 (n = 22) and 2 (n = 5) of Subulicystidium perlongisporum based on ITS nc rDNA sequences
Between the clades 1 and 2 of S. perlongisporum, there were no differences in the mean length, width and length-to-width ratio of spores (Supplementary fig. S4.1a and table S4.2). The mean length of cystidia per specimen was significantly higher in clade 2 compared with that in clade 1 (Wilcoxon W = 18, p = 0.019, Fig. 4). There were no differences in the mean width and length-to-width ratio of cystidia between clades 1 and 2.

Variation in length of cystidia within clades 1 (n = 23) and 2 (n = 5) of Subulicystidium perlongisporum. Boxes display 25th and 75th quantiles of the data range and contain a median (50% quantile). Whiskers represent 1.5 times the interquartile range and circles with a dot in the centre are the outliers
Morphological differences between long-spored Subulicystidium species
Morphological comparison based only on specimens for which DNA sequences were obtained showed that S. cochleum, S. longisporum and S. perlongisporum differed in basidiospore length (Fig. 5a). Basidiospore width and length-to-width ratio distinguished S. longisporum from S. perlongisporum and S. cochleum but did not separate the two latter species. S. perlongisporum had shorter and narrower cystidia than the two other species (Fig. 5b).

Mean size range of spores (a) and cystidia (b) in the long-spored species of Subulicystidium. Boxes (with median inside) delimit the range between 25 and 75% data quantiles (i.e. mean measurements per specimen), while higher or lower values are covered by the whiskers
Discussion
In this study, we assessed the range of molecular (nc rDNA) and morphological variation in the long-spored species of Subulicystidium: S. cochleum, S. longisporum and S. perlongisporum. We found that each of these species was monophyletic. However, none of them was sister to any of the other. For S. cochleum and S. perlongisporum, we confirmed a transoceanic distribution pattern.
We included the holotype of S. perlongisporum in our study (LY11631) and successfully obtained ITS and 28S DNA sequences from this 35-year-old specimen. We generated first DNA sequence data for the species S. cochleum. We successfully sequenced collections made in Costa Rica, Jamaica and Madagascar, but failed to obtain sequences from those made in Réunion and Zimbabwe. We provided additional morphometric and genetic data for S. longisporum. Furthermore, phylogenetic analyses allowed us to re-identify the specimen from Papua New Guinea (TU110416) as S. tropicum (previous identification: S. brachysporum). The new identification was added to the specimen and DNA sequence records in the PlutoF platform (https://plutof.ut.ee/#/specimen/view/651339; Abarenkov et al. 2010).
With the support of molecular data, we demonstrated a rather broad intraspecific variation in studied Subulicystidium species, especially in the length of spores and cystidia. Following the recommendations of Parmasto et al. (1987), we provided more accurate morphological data for S. perlongisporum, S. cochleum and S. longisporum by separating intra-individual, intraspecific and interspecific size variation. Our data showed that basidiospores of S. cochleum and S. perlongisporum can be considerably shorter than stated in the protologues of these species (Punugu et al. 1980; Boidin and Gilles 1988). Previous authors also faced this problem and apparently attributed more importance to the values of spore width and length-to-width ratio for identification (Boidin and Gilles 1988; Duhem and Michel 2001). We confirm that spore length is more variable than the spore width or length-to-width ratio in the studied species of Subulicystidium.
Despite the similarity in the shape and size of spores, the three long-spored Subulicystidium species were not closely related. Therefore, the spore shape can be considered a homoplasic character in this genus. On the other hand, we recovered a sister relationship for S. cochleum and S. acerosum. They share cystidia that are sheathed by needle-like crystals in the middle part. This crystal arrangement is different from most species in Subulicystidium that have rectangular crystals arranged in longitudinal rows. In S. oberwinkleri, a peculiar cystidium ornamentation (crystal plates) correlates with an isolated phylogenetic position. The cystidial encrustation deserves more attention in further studies on Subulicystidium.
We found that S. perlongisporum includes two sympatric lineages. Morphologically these lineages differed slightly, but significantly, in the mean length of cystidia (longer in clade 2) but not in other characters of cystidia and spores. The genetic distance between ITS nrDNA sequences from these two lineages was as high as are usually the distances between separate species within the same fungal genus, i.e. well over 3% (Schoch et al. 2012; Kõljalg et al. 2013).
Clades 1 and 2 of S. perlongisporum differed strongly in the number of available specimens, viz. 22 versus 5. The holotype of S. perlongisporum from La Réunion Island (LY 11631) was recovered in clade 1. The DNA sequences within clade 1 were more similar and were connected by shorter branches on the phylogenetic tree than the members of clade 2. We see a need for additional data for clade 2, in order to test whether it represents more than one cryptic lineage, and in order to confirm with a more balanced sampling whether the observed difference in the length of cystidia compared to clade 1 is real. Until then, we are reluctant to introduce any new species name.
Using nc ITS rDNA data, other authors showed that a single corticioid species may represent numerous phylogenetic lineages. Allopatric speciation was found within Hyphoderma setigerum and Xylodon raduloides species complexes (Nilsson et al. 2003; Fernández-López et al. 2019). In contrast, the clades of Peniophorella praetermissa species complex contained specimens of very distant geographic origin suggesting sympatric speciation (Hallenberg et al. 2007). Morphological differences between the members of the clades in the above-mentioned species complexes were often missing. Our finding for S. perlongisporum is congruent with the pattern for P. praetermissa, although the former includes only two clades while the latter has eight. It remains a challenge to explain why allopatric speciation prevails in some species complexes while genetically deviating populations may occur sympatrically in others, even when all share a saprotrophic lifestyle and live in strongly decayed wood.
Struck et al. (2018) consolidated the concept of cryptic species and stated that crypsis may represent a substantial fraction of biodiversity. They emphasized the need for a quantitative assessment of morphological disparity versus genetic divergence and comparing them with those for non-cryptic taxa. However, methods for quantitative assessment of morphological variation should become more standardized. Our protocols for morphometric analysis used for Subulicystidium (Ordynets et al. 2018; Ordynets and Denecke 2018) can be applied to all other fungi. We hope these protocols will enhance the reproducibility of the morphometric analysis in mycology and facilitate the correlation of morphometric data with genomic-scale DNA data.
References
Abarenkov K, Tedersoo L, Nilsson RH et al (2010) PlutoF—a web based workbench for ecological and taxonomic research, with an online implementation for fungal its sequences. Evol Bioinforma 6:EBO.S6271. https://doi.org/10.4137/EBO.S6271
Bengtsson-Palme J, Ryberg M, Hartmann M et al (2013) Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 4:914–919. https://doi.org/10.1111/2041-210X.12073
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, Sayers EW (2018) GenBank. Nucleic Acids Res 46(D1):D41–D47. https://doi.org/10.1093/nar/gkx1094
Boidin J, Gilles G (1988) Basidiomycètes aphyllophorales de l’Ile de la Réunion. XII: Le genre Subulicystidium Parmasto Bull Trimest la Société Mycol Fr 104:191–198
Crane S (2013) labelNodeSupport. R function. https://github.com/samuelcrane/label-node-support/blob/cf4671dc01eab90e9c7d06aae29fabae2df0f834/labelNodeSupport.r. Accessed 15 Aug 2019
Duhem B, Michel H (2001) Contribution à la connaissance du genre Subulicystidium Parmasto 1968 (Basidiomycota, Xenasmatales). Cryptogam Mycol 22:163–173. https://doi.org/10.1016/S0181-1584(01)01067-3
Fernández-López J, Telleria MT, Dueñas M et al (2019) Addressing the diversity of Xylodon raduloides complex through integrative taxonomy. IMA Fungus 10:9. https://doi.org/10.1186/s43008-019-0010-x
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes, application to the identification of mycorrihiza and rusts. Mol Ecol 2:113–118. https://doi.org/10.1111/J.1365-294x.1993.Tb00005.X
Gorjón SP, Greslebin AG, Rajchenberg M (2012) Subulicystidium curvisporum sp. nov. (Hymenochaetales, Basidiomycota) from the Patagonian Andes. Mycotaxon 118:47–52. https://doi.org/10.5248/118.47
Guindon S, Dufayard JF, Lefort V et al (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010
Hallenberg N, Nilsson RH, Antonelli A et al (2007) The Peniophorella praetermissa species complex (Basidiomycota). Mycol Res 111:1366–1376. https://doi.org/10.1016/j.mycres.2007.10.001
Harmon LJ, Weir JT, Brock CD et al (2008) GEIGER: investigating evolutionary radiations. Bioinformatics 24:129–131. https://doi.org/10.1093/bioinformatics/btm538
Harrell Jr FE, with contributions from Charles Dupont, many others. (2019) Hmisc: Harrell miscellaneous. R package version 4.2-0.https://cran.r-project.org/package=Hmisc
Henry L, Wickham H, Chang W (2019) ggstance: horizontal “ggplot2” components. R package version 0.3.2. https://cran.r-project.org/package=ggstance
Hopple JS, Vilgalys R (1999) Phylogenetic relationships in the mushroom genus coprinus and dark-spored allies based on sequence data from the nuclear gene coding for the large ribosomal subunit rna: divergent domains, outgroups, and monophyly. Mol Phylogenet Evol 13:1–19. https://doi.org/10.1006/mpev.1999.0634
Index Fungorum (2019) Index Fungorum. http://www.indexfungorum.org. Accessed 9 Oct 2019.
Izumitsu K, Hatoh K, Sumita T et al (2012) Rapid and simple preparation of mushroom DNA directly from colonies and fruiting bodies for PCR. Mycoscience 53:396–401. https://doi.org/10.1007/s10267-012-0182-3
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Katoh K, Rozewicki J, Yamada KD (2017) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. https://doi.org/10.1093/bib/bbx108
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kõljalg U, Nilsson RH, Abarenkov K et al (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277. https://doi.org/10.1111/mec.12481
Langer E (2001) Phylogeny of non-gilled and gilled basidiomycetes: DNA sequence inference, ultrastructure and comparative morphology. Habilitationsschrift, Universität Tübingen, Tübingen
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. https://doi.org/10.1093/bioinformatics/btu531
Lemoine F, Correia D, Lefort V et al (2019) NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res 47:W260–W265. https://doi.org/10.1093/nar/gkz303
Liberta AE (1980) Notes on the genus Subulicystidium. Mycotaxon 10:409–412
Liu S-L, Ma H-X, He S-H, Dai Y-C (2019) Four new corticioid species in Trechisporales (Basidiomycota) from East Asia and notes on phylogeny of the order. MycoKeys 48:97–113. https://doi.org/10.3897/mycokeys.48.31956
Maddison WP, Maddison DR (2018) Mesquite: a modular system for evolutionary analysis. Version 3.6 (build 917). http://www.mesquiteproject.org
Martini E (2020) Crusts and jells. URL: https://www.aphyllo.net, access date: 26 Feb 2020.
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: 2010 Gateway Computing Environments Workshop (GCE). IEEE, pp. 1–8.
Nilsson RH, Hallenberg N, Nordén B et al (2003) Phylogeography of Hyphoderma setigerum (Basidiomycota) in the Northern Hemisphere. Mycol Res 107:645–652. https://doi.org/10.1017/S0953756203007925
Nilsson RH, Larsson K-H, Taylor AFS et al (2018) The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1022
O’Donnell K (1992) Fusarium and its near relatives. The fungal holomorph: mitotic, meiotic and pleomorphic speciation in fungal systematics. CAB International, Wallingford, pp 225–233
Oberwinkler F (1977) Species and generic concepts in the Corticiaceae. In: The species concept in Hymenomycetes. Cramer, Vaduz, pp. 331–348.
Ordynets A (2018) Plotting phylogenetic tree with branch supports from two phylogenetic analyses. In: protocols.io. https://doi.org/10.17504/protocols.io.n9fdh3n
Ordynets A, Denecke J (2018) Calculating and plotting size range of morphological structures. In: protocols.io. https://doi.org/10.17504/protocols.io.n7tdhnn
Ordynets A, Scherf D, Pansegrau F et al (2018) Short-spored Subulicystidium (Trechisporales, Basidiomycota): high morphological diversity and only partly clear species boundaries. MycoKeys 35:41–99. https://doi.org/10.3897/mycokeys.35.25678
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. https://doi.org/10.1093/bioinformatics/btg412
Parmasto E, Parmasto I, Möls T (1987) Variation of basidiospores in the hymenomycetes and its significance to their taxonomy. J. Cramer, Berlin, Stuttgart
Patouillard NT (1894) Quelques espèces nouvelles de Champignons du nord de l’Afrique. J Bot 8:219–221
Piel W, Chan L, Dominus M, et al. (2009) TreeBASE v. 2: a database of phylogenetic knowledge. In: e-BioSphere 2009. London.
Punugu A, Dunn MT, Welden AL (1980) Peniophoroid fungi of the West Indies. Mycotaxon 10:428–454
R Core Team (2019) R: a language and environment for statistical computing. Version 3.5.3https://www.r-project.org/
Rambaut A (2014) FigTree, a graphical viewer of phylogenetic trees. Version 1.4.4. http://tree.bio.ed.ac.uk/software/figtree/
Ronquist F, Teslenko M, van der Mark P et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Rüdigs J (2019) Makroaufmaßprogramm. URL: https://ruedig.de/tmp/messprogramm.htm. Accessed 15 Aug 2019.
Schoch CL, Seifert KA, Huhndorf S et al (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci 109:6241–6246. https://doi.org/10.1073/pnas.1117018109
Struck TH, Feder JL, Bendiksby M et al (2018) Finding evolutionary processes hidden in cryptic species. Trends Ecol Evol 33:153–163. https://doi.org/10.1016/j.tree.2017.11.007
Tedersoo L, Jairus T, Horton BM et al (2008) Strong host preference of ectomycorrhizal fungi in a Tasmanian wet sclerophyll forest as revealed by DNA barcoding and taxon-specific primers. New Phytol 180:479–490. https://doi.org/10.1111/j.1469-8137.2008.02561.x
Vaidya G, Lohman DJ, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27:171–180. https://doi.org/10.1111/j.1096-0031.2010.00329.x
Volobuev S (2016) Subulicystidium perlongisporum (Trechisporales, Basidiomycota) new to Russia, with notes on a molecular study of the species. Nov Hedwigia 102:531–537. https://doi.org/10.1127/nova_hedwigia/2016/0329
Warnes GR, Bolker B, Gorjanc G, et al. (2017) gdata: various R programming tools for data manipulation. R package version 2.18.0. https://cran.r-project.org/package=gdata
White TJ, Bruns S, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand D, Sninsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications. Academic Press, New York, pp 315–322
Wickham H (2009) ggplot2: elegant graphics for data analysis. Springer, New York
Wickham H (2019) forcats: tools for working with categorical variables (factors). R package version 0.4.0. https://cran.r-project.org/package=forcats
Wickham H, François R, Henry L, Müller K (2019) dplyr: a grammar of data manipulation. R package version 0.8.0. https://cran.r-project.org/package=dplyr
Wilk J (2012) Smaff - “Statistische Messreihen-Auswertung für Fungi v3.1.”. Südwestdeutsche Pilzrundschau 48:49–56
Wright K (2018) pals: color palettes, colormaps, and tools to evaluate them. R package version 1.5. https://cran.r-project.org/package=pals
Yu G (2019) treeio: base classes and functions for phylogenetic tree input and output. R package version 1.6.2. https://guangchuangyu.github.io/software/treeio
Yu G, Smith DK, Zhu H et al (2017) ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8:28–36. https://doi.org/10.1111/2041-210X.12628
Acknowledgements
We would like to thank the curators of herbaria and mycologists who provided loan specimens for our study: Ibai Olariaga (ARAN), Markus Scholler (KR), Mélanie Thiébaut (LY), Irja Saar (TU), Cornelia Dilger-Endrulat and Franz Oberwinkler (TUB). Ulrike Frieling and Sylvia Heinemann (University of Kassel) and Rasmus Puusepp (University of Tartu) are deeply acknowledged for the assistance with the molecular laboratory work. We thank the reviewers and editors for their comments that helped to improve the manuscript.
Funding
Open Access funding provided by Projekt DEAL. The work of A. Ordynets and E. Langer was partly funded by the LOEWE excellence initiative of the state of Hesse (Germany) within the framework of the Cluster for Integrative Fungal Research (IPF). K.-H. Larsson received support from SIU, Norwegian Centre for International Cooperation in Education (projects CAPES-SIU-2013/10057 and CAPES-SIU-2015/10004). The work of S. Volobuev and S. Bolshakov was carried out within the framework of the institutional research project АААА-А19-119020890079-6 using equipment of The Core Facilities Center “Cell and Molecular Technologies in Plant Science” at the Komarov Botanical Institute RAS (St.-Petersburg, Russia).
Author information
Authors and Affiliations
Contributions
A. Ordynets conceived the idea. K.-H. Larsson, E. Langer, A. Saitta, S. Volobuev, S. Bolshakov, B. Buyck and E. Yurchenko collected and identified fungal specimens. R. Liebisch, A. Ordynets, A. Saitta, D. Scherf, L. Lysenko, S. Volobuev and S. Bolshakov obtained morphometric data. R. Liebisch, A. Saitta, D. Scherf, L. Lysenko, S. Volobuev, S. Bolshakov and K.-H. Larsson performed molecular lab work and generated DNA sequences. A. Ordynets, R. Liebisch and D. Scherf analyzed the data. A. Ordynets wrote the first version of the manuscript and all authors read, contributed to and agreed on the final version.
Corresponding author
Additional information
Section Editor: Yu-Cheng Dai
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Basidiomycote Mycology in honor of Franz Oberwinkler who passed away in March 2018.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ordynets, A., Liebisch, R., Lysenko, L. et al. Morphologically similar but not closely related: the long-spored species of Subulicystidium (Trechisporales, Basidiomycota). Mycol Progress 19, 691–703 (2020). https://doi.org/10.1007/s11557-020-01587-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-020-01587-3