
Abstract
Knowledge of genetic diversity in A. rabiei provides different levels of information that are important in the management of crop germplasm resources. Gene flow on a regional level indicates a significant potential risk for the regional spread of novel alleles that might contribute to fungicide resistance or the breakdown of resistance genes. Simple sequence repeat (SSR) and mating type (MAT) markers were used to determine the genetic structure, and estimate genetic diversity and the prevalence of mating types in 103 Ascochyta rabiei isolates from seven counties in the Ilam and Kermanshah provinces of western Iran (Ilam, Aseman abad, Holaylan, Chardavol, Dareh shahr, Gilangharb, and Sarpul). A set of 3 microsatellite primer pairs revealed a total of 75 alleles; the number of alleles varied from 15 to 34 for each marker. A high level of genetic variability was observed among A. rabiei isolates in the region. Genetic diversity was high (He = 0.788) within populations with corresponding high average gene flow and low genetic distances between populations. The smallest genetic distance was observed between isolates from Ilam and Chardavol. Both mating types were present in all populations, with the majority of the isolates belonging to Mat1-1 (64%), but within populations the proportions of each mating type were not significantly different from 50%. Results from this study will be useful in breeding for Ascochyta blight-resistant cultivars and developing necessary control measures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ascochyta blight, caused by the fungus Ascochyta rabiei (Pass.) Labr., is the most destructive disease of aerial parts of chickpea (Cicer arietinum L.) in chickpea-producing regions of the world (Nene and Reddy 1987). Iran is the fourth largest chickpea producer in the world (http://faostat.fao.org) of which Kermanshah and Ilam are important chickpea growing areas that suffer severe epidemics every year. Ascochyta rabiei is a haploid, heterothallic dothideomycete fungus within the class Pleosporales (Trapero-Casas and Kaiser 1992; Wilson and Kaiser 1995). The life cycle of this fungus involves several asexual generations during the growing season of the crop, and a sexual or perfect stage (Didymella rabiei (Kovachevski) v. Arx.) which develops on infected plant debris during the winter (Navas-Cortes et al. 1998; Trapero-Casas and Kaiser 1992; Trapero-Casas et al. 1996). Although the teleomorph’s influence on disease epidemiology and pathogen diversity is still unclear, it may be more widespread than previously thought, and ascospores may serve as a primary inoculum (Kaiser and Hannan 1987; Trapero-Casas et al. 1996).
The disease appears in the field firstly as small infected foci and then spreads quickly when cool and wet conditions prevail. The severity of disease can be reduced through the use of resistant cultivars, fungicide applications, and cultural practices such as crop rotation, removal of debris, and sowing clean seed. Periodic appearances of new and more virulent strains of the pathogen are a major problem in resistance breeding (Grewal 1984; Singh et al. 1981).
The genetic structure of a population is the amount and distribution of genetic variation within and among populations of a species and reflects the evolutionary history of populations (McDonald and Linde 2002). The amount of genetic variation within a population indicates how rapidly a pathogen can evolve and thus provides valuable information in predicting the effectiveness of control measures such as host resistance genes or chemical control (McDermott and McDonald 1993). The main evolutionary forces that contribute to the genetic structure within and between populations are gene flow, genetic drift, reproduction/mating systems, population size, and selection. Molecular markers have been widely deployed to detect and identify A. rabiei isolates and to examine genetic diversity, genetic structure, and virulence in populations of this fungus (Morjane et al. 1994; Peever et al. 2004; Phan et al. 2003; Santra et al. 2001; Udupa et al. 1998). Significant genetic variation was shown in populations of A. rabiei in Italy (Fischer et al. 1995), Tunisia (Geistlinger et al. 2000; Morjane et al. 1994), Syria and Lebanon (Udupa et al. 1998), Spain (Navas-Cortés et al. 1998), Pakistan (Jamil et al. 2000) and India (Santra et al. 2001). In some regions of the world where chickpea has only recently been introduced, for example Australia and California, only modest genetic diversity and a single mating type has been reported (Kaiser 1997; Phan et al. 2003).
To examine genetic diversity and population structure among 103 A. rabiei isolates from western Iran, we deployed simple sequence repeat (SSR) markers developed by Geistlinger et al. (2000). Amongst different genetic markers available for such studies, SSRs are routinely used because they are abundant, distributed over the euchromatic part of the genome and are highly polymorphic (Schlötterer and Ellegren 1998; Zane et al. 2002). The presence, distribution and frequency of A. rabiei mating types in regions of Tunisia have been used to shed light on recent introductions as opposed to stable, randomly mating populations (Rhaiem et al. 2007). To examine the relative proportions of MAT 1-1or MAT 1-2 allelic groups in this study, we applied the MAT-specific multiplex PCR assay developed by Barve et al. (2003).
Materials and methods
Sampling, isolation and maintenance of fungal isolates
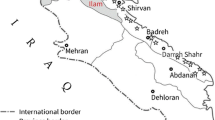
Chickpea plants with brown or black lesions on stem, leaves and pods were randomly collected in 2007 from seven different regions in the western Iranian Ilam and Kermanshah provinces. For the purposes of this study, isolates belonging to these regions are considered as populations (Fig. 1; Table 1). While these regions are geographically juxtaposed, they vary in altitude and climate, are separated by substantial mountain ranges (largely but not exclusively along a north-west to south-east axis) and most rural areas have limited road access. Diseased samples were cut into 2- to 5-mm-long pieces, surface sterilized by dipping into domestic bleach solution (1% NaOCl) for 2–3 min, washed three times with sterile distilled water, dried with sterile filter paper and plated on potato dextrose agar (PDA) or chickpea seed meal agar (CSDA). Samples were incubated for 3 days in an incubator at 23°C with a 12-h photoperiod to induce production of conidia. Isolates were single-spored by harvesting conidia from a single pycnidium with a sterile needle and streaking them on a 2% water agar plate. Conidia were incubated as above and a single germinated conidium sub-cultured onto PDA or CSDA plates and incubated with a 12-h photoperiod for 2–3 weeks. Single-conidial isolates were stored on sterile filter paper at 20°C.

Geographical origins of the seven A. rabiei populations in western Iran used in this study
DNA extraction
Liquid cultures were initiated by adding 2–4 mm2 pieces of filter paper to 250-mL Erlenmeyer flasks containing 100 mL PDB medium (potato dextrose broth plus 2 g yeast extract per liter). Flasks were incubated at room temperature approximately 25°C on a rotary shaker for 6–8 days. Mycelium was collected by filtration through sterile filter paper with a vacuum funnel. Mycelia were harvested, frozen and stored at −20°C. DNA was extracted using a modified hexadecyl trimethyl-ammonium bromide (CTAB) procedure (Doyle and Doyle 1987). Mycelia were ground in liquid nitrogen and suspended in 2% CTAB extraction buffer (1.4 M NaCl, 0.1 M Tris-HCl, pH 8.0, 20 mM EDTA, 0.2% β-mercaptoethanol). Samples were treated with 5 units RNAse at 37°C for 30 min., then extracted with chloroform-isoamyl alcohol 24:1 (v/v). DNA in the supernatant was precipitated with isopropanol, rinsed with ethanol, and adjusted to a final concentration of 20 ng/μl in TE (pH 7.4).
SSRs and mating type (MAT) amplification and analysis
We selected three SSR primer pairs (Table 2) on the basis of their high PIC as described by Geistlinger et al. (2000). SSRs were amplified using the Multiplex Ready Technique (Hayden et al. 2008). The optimal concentration of locus-specific primer required in the multiplexed PCR reactions was determined as 10 nM for each primer. Fragment analysis for visualization of SSRs was performed on ABI3730 DNA analyzer (Applied Biosystems) and SSR allele sizing and calling was performed using GeneMapper v. 3.7 (Applied Biosystems). To determine mating types of the isolates, PCR with the allele specific primers Com1, Tail5, and SP21 was performed as described by Barve et al. (2003). PCR products were separated electrophoretically on 1.5% agarose gels.
Statistical analysis
To characterize genetic variation, observed number of alleles (Na), effective number of alleles (Ne), Nei's gene diversity (He), and Shannon's information index (I) were calculated for each locus and population. All calculations were performed using POPGENE program v. 1.31 (Yeh et al. 1999). Mean values of gene diversity of total populations (Ht), gene diversity between populations (Hs), proportion of gene diversity attributable to differentiation among populations (Gst), and estimates of gene flow from Gst (Nm) were obtained across loci (McDermott and McDonald 1993). A dendrogram of genetic relationships between populations was estimated from the SSR data using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) clustering method on the basis of Nei’s (1978) unbiased genetic distance.
Results
Distribution of alleles at polymorphic SSR loci
All three microsatellite loci were highly informative with a total of 75 alleles detected between them. ArH06T, ArA03T, and ArH05T showed a total of 26, 34, and 15 alleles per locus, respectively. The average number of alleles per locus was the highest in the two populations of Chardavol and Dareh shahr (9.33 alleles) and the lowest in the Holylan population (5.33) (Table 3). A summary of the genetic diversity data of three microsatellite locus is given in Table 4. Gene diversity (He) and Shannon's information index (I) were the highest in the locus ArA03T (He = 0.9; I = 2.94) while the lowest values were estimated for the locus ArH05T (He = 0.67; I = 1.77).
Genetic variability of population
Genetic parameters for the three microsatellite loci in the seven A. rabiei populations are given in Table 5. Observed (Na) and effective (Ne) numbers of alleles were higher in Chardavol and Dareh shahr compared to other populations. The gene diversity (He) and the Shannon's information index (I) indices were also higher in Dareh shahr, but lower in Holaylan. Based on the SSR data, the average genetic distance was calculated among the seven populations. The lowest genetic distance was found between Chardavol and Sarpul, while the highest genetic distance was revealed between Ilam and Darah shahr (Table 6).
The total gene diversity (Ht) and gene diversities between subpopulations (Hs) were estimated to be 0.78 and 0.73, respectively. Moreover, gene diversity attributable to differentiation among populations (Gst) was 0.07, while gene flow (Nm) was 6.64.
Genetic relationships among populations and distribution of mating types
Nei’s pairwise genetic distances between the populations varied from 0.15 to 0.36. Cluster analysis (UPGMA) was used to produce a dendrogram showing the genetic relationships between the populations based on the SSR data (Fig. 2). The lowest genetic distance was between Chardavol and Sarpul and then between Aseman abad and Holaylan. Moreover, the dendrogram showed a distinction between the Dareh shahr population and the six remaining populations.

Dendrogram of genetic relationships between seven populations of A. rabiei as reconstructed by UPGMA using Nei’s genetic distance
Both mating types were found in all seven populations and the frequencies of mating types were variable between populations. The majority of the isolates belonged to Mat1-1 (64.08%) with the remainder (35.92%) being Mat1-2, but no population was significantly different from a 50:50 ratio.
Discussion
This study reveals for the first time the levels of genotypic diversity among A. rabiei isolates occurring in the provinces of Ilam and Kermanshah in western Iran. The region sampled comprised seven counties over a maximum distance of 350 km. In comparison to other studies using the same SSRs markers (Geistlinger et al. 2000; Rhaiem et al. 2008; Varshney et al. 2009), this area may represent the highest level of allelic diversity detected to date. Although sample sizes are not comparable, at locus ArH06 over similar or larger areas, those studies identified 2, 9, and 15 alleles, respectively, compared to 26 here. Geographically, the SSR data indicated that all seven populations had high levels of gene diversity, with Dareh shahr and Aseman abad showing the highest diversity. This is perhaps unsurprising given western Iran’s central position in the original geographic range of chickpea, which comprises the Fertile Crescent and Asia Minor (van der Maesen 1987).
Over 93% of the diversity in this study was distributed on a local level within populations and correspondingly a low level of gene diversity (Gst = 0.07) was detected among all seven populations. This indicates the majority of variation is within populations with low genetic differentiation among the populations and little evidence for geographical subdivision. Individuals within such populations are likely to be genetically different, with each population containing the same or similar complement of alleles in similar frequencies. This compares to Tunisia where 123 A. rabiei isolates representative of five regions were analyzed to estimate genetic diversity and where a high level of genetic differentiation was detected among subpopulations (Gst = 0.33; Rhaiem et al. 2007). In contrast, on a smaller scale in Tunisia, Morjane et al. (1994) examined the genetic variation among 50 isolates sampled from one single chickpea field. Most of the genetic variability was attributable to diversity within locations rather than between locations. In Australia, a study based on STMS fingerprints found none of the alleles showed significant differences in allele frequencies, and a very low level of gene diversity (Ht = 0.02) was found with the majority of the diversity (92%) distributed within subpopulations. This reflects a relatively recent introduction of a single mating type of A. rabiei with modest genetic variability (Phan et al. 2003).
Gene flow is one of the evolutionary forces that can have a significant force on the genetic diversity of a population. In the absence of gene flow, genetic drift causes different allele frequencies at neutral loci, leading to differentiation in isolate populations (Keller et al. 1997). The low genetic differentiation in this study has several possible explanations. Geographic distances between populations are modest, allowing movement of isolates through infected plant debris, infected seeds, and agricultural vehicles. The exchange of chickpea seeds is possibly the commonest cause, because farmers in the region often purchase seed from other farmers. This undermines practices that that might be expected to promote population differentiation, such as the sale of certified seed from central stores in each county and the propagation of seed by some farmers for their own use. Other factors affecting selection and population differentiation include the deployment of different cultivars and fungicides. In western Iran, the number of cultivars is limited to about six Kabuli types, occasionally augmented by resistant cultivars by agricultural research centers, together with a local Kabuli-Desi mixture propagated by individual farmers. Fungicide use is limited or absent.
On the assumption that the high degree of population similarity is due to gene flow and that the populations are at equilibrium, we can use GST to estimate Nm, the average number of migrants that would need to be exchanged among populations in each generation to account for their present lack of subdivision. Nm averaged 6.64 in all loci and populations, suggesting a level of gene flow that was 6 times greater than needed to prevent populations from diverging by genetic drift (Keller et al. 1997). This result may help explain why resistance in new cultivars, periodically released by agricultural research centers and including material from the International Centre for Agricultural Research in the Dry Areas (INCARDA), has proven ineffective after 2–3 years (data not shown).
The results also showed different allele frequencies between the SSR loci and populations, with isolates from Dareh shahr, the eastern most county, appearing most different. Farmers in the region usually cultivate cereals in rotation with chickpea and the stubble is either burnt or grazed by livestock. However, chickpea debris may also be ploughed in after harvesting, only a limited number of cultivars are farmed, sequential cultivation of more than one chickpea crop occurs in regions with mild climates, and infected seed is exchanged locally between farmers. These practices would promote the asexual spread of A. rabiei asexually, which in combination with these cultivation practices may contribute to different allele frequencies. The sample sizes per population and number of SSRs primers were too small in this experiment for a statistically powerful test of gametic equilibrium.
Moreover, infected seed can lead to persistence of genotypes and may explain the different allele frequencies diversity of genotypes found at the end of the growing season in natural populations. If the main source of primary inoculum was asexual and from seeds, we might expect to find some clones that were distributed among different locations in a field. After a while, genetic drift and selection would limit the number of genotypes present in field populations, unless sexual reproduction generated new genotypes and wind dispersal distributed ascospores among populations every year (Shah et al. 1995). Pycnidiospores are likely to play an important role in epidemiology over small distances (1–2 m), but ascospores probably play a more important role in the population biology and perhaps in the epidemiology of disease on a regional scale (tens to hundreds of kilometers; Keller et al. 1997).
The high genetic diversity of A. rabiei detected in this study has important implications for farming practices and breeding efforts in the area. Gene flow in the region suggests a significant potential risk for the spread of novel alleles that might contribute to fungicide resistance or the breakdown of resistance genes. Approaches such as using different combinations of pyramided resistance genes or mixtures of cultivars may offer more durable resistance.
References
Barve MP, Arie T, Salimath SS, Muehlbauer FJ, Peever TL (2003) Cloning and characterization of the mating type (MAT) locus from Ascochyta rabiei (teleomorph: Didymella rabiei) and a MAT phylogeny of legume-associated Ascochyta spp. Fungal Genet Biol 39:151–167
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Fischer C, Porta-Puglia A, Barz W (1995) RAPD Analysis of Pathogenic Variability in Ascochyta rabiei. J Phytopathol 143:601–607
Geistlinger J, Weising K, Winter P, Kahl G (2000) Locus-specific microsatellite markers for the fungal chickpea pathogen Didymella rabiei (anamorph) Ascochyta rabiei. Mol Ecol 9:1939–1941
Grewal JS (1984) Evidence of physiological races in Ascochyta rabiei of chickpea. In: Saxena MC, Singh KB (eds) Ascochyta blight and winter sowing of chickpea. Nijhoff/Junk, The Hague, pp 55–65
Hayden M, Nguyen T, Waterman A, Chalmers K (2008) Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping. BMC Genomics 9:80
Jamil FF, Sarwar N, Sarwar M, Khan JA, Geistlinger J, Kahl G (2000) Genetic and pathogenic diversity within Ascochyta rabiei (Pass.) Lab. populations in Pakistan causing blight of chickpea (Cicer arietinum L.). Physiol Mol Plant Pathol 57:243–254
Kaiser WJ (1997) Inter—and intranational spread of ascochyta pathogens of chickpea, faba bean, and lentil. Can J Plant Path 19:215–244
Kaiser WJ, Hannan RM (1987) First report of Mycosphaerella rabiei on chickpeas in the Western Hemisphere. Plant Dis 71:192
Keller SM, McDermott JM, Pettway RE, Wolfe MS, McDonald BA (1997) Gene flow and sexual reproduction in the wheat glume blotch pathogen Phaeosphaeria nodorum (anamorph Stagonospora nodorum). Phytopathology 87:353–358
McDermott JM, McDonald BA (1993) Gene flow in plant pathosystems. Annu Rev Phytopathol 31:353–373
McDonald BA, Linde C (2002) The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica 124:163–180
Morjane H, Geistlinger J, Harrabi M, Weising K, Kahl G (1994) Oligonucleotide fingerprinting detects genetic diversity among Ascochyta rabiei isolates from a single chickpea field in Tunisia. Curr Genet 26:191–197
Navas-Cortés J, Pérez-Artés E, Jiménez-Diaz R, Llobell A, Bainbridge B, Heale J (1998) Mating type, pathotype and RAPDs analysis in Didymella rabiei, the agent of ascochyta blight of chickpea. Phytoparasitica 26:199–212
Navas-Cortes JA, Trapero-Casas A, Jimenez-Diaz RM (1998) Phenology of Didymella rabiei development on chickpea debris under field conditions in Spain. Phytopathology 88:983–991
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nene YL, Reddy MV (1987) Chickpea diseases and their control. In: Sexena MK, Singh KB, Johansen C (eds) The chickpea. CABI International, Wallingford, pp 233–270
Peever TL, Salimath SS, Su G, Kaiser WJ, Muehlbauer FJ (2004) Historical and contemporary multilocus population structure of Ascochyta rabiei (teleomorph: Didymella rabiei) in the Pacific Northwest of the United States. Mol Ecol 13:291–309
Phan HTT, Ford R, Taylor PWJ (2003) Population structure of Ascochyta rabiei in Australia based on STMS fingerprints. Fungal Divers 13:111–129
Rhaiem A, Cherif M, Dyer PS, Peever TL (2007) Distribution of mating types and genetic diversity of Ascochyta rabiei populations in Tunisia revealed by mating-type-specific PCR and random amplified polymorphic DNA markers. J Phytopathol 155:596–605
Rhaiem A, Chérif M, Peever TL, Dyer PS (2008) Population structure and mating system of Ascochyta rabiei in Tunisia: evidence for the recent introduction of mating type 2. Plant Pathol 57:540–551
Santra DK, Singh G, Kaiser WJ, Gupta VS, Ranjekar PK, Muehlbauer FJ (2001) Molecular analysis of Ascochyta rabiei (Pass.) Labr., the pathogen of ascochyta blight in chickpea. Theor Appl Genet 102:676–682
Schlötterer C, Ellegren H (1998) Genome evolution: Are microsatellites really simple sequences? Curr Biol 8:132–134
Shah D, Bergstrom GC, Ueng PP (1995) Initiation of septoria nodorum blotch epidemics in winter wheat by seedborne Stagonospora nodorum. Phytopathology 85:452–457
Singh KB, Hawtin GC, Nene YL, Reddy MV (1981) Resistance in chickpeas to Ascochyta rabiei. Plant Dis 65:586–587
Trapero-Casas A, Kaiser WJ (1992) Development of Didymella rabiei, the teleomorph of Ascochyta rabiei, on chickpea straw. Phytopathology 82:1261–1266
Trapero-Casas A, Navas-Cortés JA, Jiménez-Díaz RM (1996) Airborne ascospores of Didymella rabiei as a major primary inoculum for Ascochyta blight epidemics in chickpea crops in southern Spain. Eur J Plant Pathol 102:237–245
Udupa SM, Weigand F, Saxena MC, Kahl G (1998) Genotyping with RAPD and microsatellite markers resolves pathotype diversity in the ascochyta blight pathogen of chickpea. Theor Appl Genet 97:299–307
van der Maesen LJG (1987) Origin, history and taxonomy of chickpea. In: Saxena MC, Singh KB (eds) The chickpea. CABI, Wallingford, pp 11–14
Varshney R, Pande S, Kannan S, Mahendar T, Sharma M, Gaur P, Hoisington D (2009) Assessment and comparison of AFLP and SSR based molecular genetic diversity in Indian isolates of Ascochyta rabiei, a causal agent of Ascochyta blight in chickpea (Cicer arietinum L.). Mycol Prog 8:87–97
Wilson AD, Kaiser WJ (1995) Cytology and genetics of sexual compatibility in Didymella rabiei. Mycologia 87:795–804
Yeh F, Yang R, Boyle T (1999) POPGENE version 1.31. Microsoft Windows-based software for population genetics analysis. University of Alberta and Centre for International Forestry Research, Alberta
Zane L, Bargelloni L, Patarnello T (2002) Strategies for microsatellite isolation: a review. Mol Ecol 11:1–16
Acknowledgements
The authors would like to thank Ms Julie Lawrence and Ms Kasia Rybak from the ACNFP for technical support. Research at the ACNFP is supported by the Australian Grains Research and Development Corporation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s11557-010-0689-y
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nourollahi, K., Javannikkhah, M., Naghavi, M.R. et al. Genetic diversity and population structure of Ascochyta rabiei from the western Iranian Ilam and Kermanshah provinces using MAT and SSR markers. Mycol Progress 10, 1–7 (2011). https://doi.org/10.1007/s11557-010-0668-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-010-0668-3