
Abstract
Background
To study the role of computed tomography (CT)-derived radiomics features and clinical characteristics on the prognosis of “driver gene-negative” lung adenocarcinoma (LUAD) and to explore the potential molecular biological which may be helpful for patients’ individual postoperative care.
Methods
A total of 180 patients with stage I-III “driver gene-negative” LUAD in the First Affiliated Hospital of Sun Yat-Sen University from September 2003 to June 2015 were retrospectively collected. The Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression model was used to screen radiomics features and calculated the Rad-score. The prediction performance of the nomogram model based on radiomics features and clinical characteristics was validated and then assessed with respect to calibration. Gene set enrichment analysis (GSEA) was used to explore the relevant biological pathways.
Results
The radiomics and the clinicopathological characteristics were combined to construct a nomogram resulted in better performance for the estimation of OS (C-index: 0.815; 95% confidence interval [CI]: 0.756–0.874) than the clinicopathological nomogram (C-index: 0.765; 95% CI: 0.692–0.837). Decision curve analysis demonstrated that in terms of clinical usefulness, the radiomics nomogram outperformed the traditional staging system and the clinicopathological nomogram. The clinical prognostic risk score of each patient was calculated based on the radiomics nomogram and divided by X-tile into high-risk (> 65.28) and low-risk (≤ 65.28) groups. GSEA results showed that the low-risk score group was directly related to amino acid metabolism, and the high-risk score group was related to immune and metabolism pathways.
Conclusions
The radiomics nomogram was promising to predict the prognosis of patients with “driver gene-negative” LUAD. The metabolism and immune-related pathways may provide new treatment orientation for this genetically unique subset of patients, which may serve as a potential tool to guide individual postoperative care for those patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the second most common malignant tumor but the most common cause of malignancy-related mortality worldwide [1]. More than 80% of histological types of lung cancer are lung adenocarcinoma (LUAD) [2]. Over the last decade, targeted therapy has greatly prolonged the overall survival of driver gene-mutated lung cancer patients [3, 4]. In addition to EGFR mutations and ALK fusions, driver genes also include KRAS, BRAF and HER2, as well as gene rearrangements involving RET, ROS1 and MET exon 14 skipping [5, 6]. However, 14–25% of LUAD patients are still “driver gene-negative” [5], which identified as patients with LUAD negative for EGFR, KRAS, BRAF, HER2, MET, ALK, RET and ROS1 [3]. And with the development of large-panel sequencing technology, this proportion is increasing. Although immune checkpoint inhibitors (ICIs) such as anti-programmed death-1 (PD-1)/PD-ligand-1 (PD-L1) have shown great potential in the treatment of lung cancer [7], the large number of non-responders and the immune-related toxicities of ICIs limits the application of immunotherapy in LUAD [8]. From the current research results, the first-line pembrolizumab monotherapy demonstrated an OS benefit than chemotherapy for patients with advanced non-small cell lung cancer who have not been treated before and without EGFR/ALK aberrations [9], whereas, for patients with “driving gene-negative” LUAD, the treatment option is limited [5] and the efficacy of ICIs still needs further exploration [9, 10].
Medical imaging technology plays an important role in clinical management. By extracting multiple quantitative features from conventional computed tomography (CT) images, radiomics could capture the differences between different tumor phenotypes non-invasively [11]. Recent advances in radiomics showed that it has provided potential usefulness personalized insights increasingly in oncologic practice such as tumor detection, subtype classification, prognosis and treatment response assessment [12,13,14]. The nomogram obtained by combining radiomics with clinicopathological characteristics appears to improve the accuracy of prognosis prediction [15]. However, the radiomic nomogram associated with the prognosis of “driver gene-negative” LUAD has not yet been described.
Therefore, the purpose of the present study was to build a nomogram based on radiomics CT features and clinical characteristics to predict the survival of patients with “driver gene-negative” LUAD and to evaluate the incremental value of radiomic signatures to traditional staging systems and clinicopathological risk factors. In addition, we intended to use radiomics nomogram and gene expression data to explore the potential molecular biological in order to guide individual postoperative care for those patients.
Materials and methods
Patients
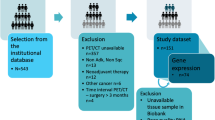
This study was approved by the Ethics Committee of The First Affiliated Hospital of Sun Yat-Sen University with a waiver of informed consent (No. [2021]531; date of approval: 20/08/2021). The data of bulk RNA-seq had been uploaded to the Gene Expression Omnibus (GEO) database by our previous published article [3]. We selected 371 “driver gene-negative” LUAD patients in the First Affiliated Hospital of Sun Yat-Sen University from September 2003 to June 2015. None of the patients underwent any antitumor therapy before biopsy sampling, and "driver gene–negative" status was determined in paraffin-embedded (FFPE) tissues. A total of 60 pairs of fresh tumor and adjacent normal tissues were selected randomly from the 371 patients with “driver gene–negative” LUAD and were used for genome-wide microarray assay to screen candidate genes followed by Western blotting and qPCR. Circulating tumor cell (CTC) enumeration and PD-L1 expression detection were performed on each eligible patient. A total of 191 patients were excluded from this study because without surgical resection and poor CT image quality.
The final patient cohort was 180 patients from June 2007 to June 2015 after screening by strict inclusion and exclusion criteria. Only 28 patients’ fresh tumors and adjacent normal tissues sequencing results were available, including 11 patients of stage I, 5 patients of stage II and 12 patients of stage III. All chest CT images with an 1-mm axial reconstruction interval were acquired within 2 weeks (median value of 6 days). And then, the training and validation data sets were assigned 7-to-3, and the latter included the 28 patients with available genetic data. The workflow is shown in Supplementary Fig. 1.
The endpoint of this study was OS, which was defined as the time from the date of pathological diagnosis to death or the last follow-up. All patients were followed up for at least five years, unless the patient died. Baseline clinical–pathological data, such as age, sex, smoking status, stage, histologic grade, were obtained from the medical records (Table 1).
PD-L1 protein expression detection and CTC enumeration
Immunohistochemical analysis of PD-L1 expression was detected with primary rabbit monoclonal antibody against human PD-L1 (SP263; 1:2000; Roche Ventana, Tucson, AZ, USA) in deparaffinized and hydrated tumor tissue. PD-L1 expression depended on the intensity of cell membrane staining. The proportion of PD-L1-positive cells was independently estimated as the percentage of total tumor cells in whole sections by two pathologists. If the independent assessments did not agree, the slides were reviewed by the two investigators together to achieve consensus. The consensus judgments were adopted as the final results. The PD-L1 expression is defined by tumor proportion scores (TPS). PD-L1 TPS ≥ 1% was defined as positive expression, and PD-L1 TPS ≥ 50% was defined as high expression.
The methodology of CTC enumeration was detailed in our previous study [16].
Image acquisition and radiomics processing
As for the acquisition parameters and retrieval procedures of CT images in our study, more details are presented in Supplementary material E1. Tumors were delineated by the junior radiologist (with 6 years of clinical experience in chest CT study interpretation) on the CT images using the same active-contour semiautomatic algorithm and verified by the expert radiologists (with 12 years of clinical experience in chest CT study interpretation) through the ITK-SNAP software (version 3.8.0, https://www.itksnap.org). Radiomics features were extracted from CT images resampled to isometric voxels of 1 × 1 × 1 mm3 by using the PyRadiomics platform [17] implemented in Python software (version 3.8.3, https://www.python.org). The details of the platform and radiomics features are described in Supplementary material E2. Totally 1409 radiomics features were, respectively, extracted from plain and enhanced CT images, including 14 shape features, 18 first-order intensity statistics features and 75 texture features (Table S1), as well as 558 Laplacian of Gaussian (LOG) features, 744 wavelet features.
Radiomics features selection and radiomics score calculation
A two-way random, single-measure (absolute agreement) intraclass correlation coefficient (ICC) was used to evaluate the robustness of the extracted radiomics features. If the ICC value was greater than 0.8 in the training data set, then the stability of the feature can be considered excellent [18].
The variables included in the multivariate analysis should follow Harrell's guidelines [19], that is, the number of events of interest should exceed the number of variables by at least 5 times. However, including more variables does not necessarily lead to higher accuracy, but leads to overfitting [20]. To further remove redundant features in the training data set, the Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression model [21, 22], a popular regularized machine learning algorithm suitable for dimensionality reduction of high-dimensional data, was used to select the most useful radiomics characteristics which related to prognosis with tenfold nested cross-validation. The selected radiomics features were then weighted by their respective coefficients and summed to get the radiomics score (Rad-score) of each patient [23].
Radiomics nomogram construction
The clinical pathological data of the training data set were analyzed by single-factor Cox regression analysis to evaluate the potential relationship between each selected feature and OS. And then, the independent clinical–pathological risk factors were selected by the multivariate Cox analysis and used to construct a clinicopathological nomogram.
Integrated the selected independent clinicopathological risk factors and the Rad-score into the Cox regression model, and construct a radiomics nomogram via the training set. In order to quantify the prognostic risk, the clinical prognostic score of patients in the validation data set was calculated by the nomogram. Calibration curves were used to compare the consistency between the observed results and the OS correlation of clinicopathological nomogram and radiomics nomogram.
As for the evaluation of gain effect whether the radiomics characteristics would have on the clinical prognosis prediction performance of the clinical pathological factor model, the Harrell Concordance Index (C-index) was calculated. The improvement of usefulness brought by the radiomics feature was quantitatively calculated using the integrated discrimination improvement (IDI) [24], which is very sensitive in detecting prediction probability changes in a new model compared to an old model. The Akaike information criteria (AIC) were used to assess the risk of overfitting [25]. And then, calculate the net income under different threshold probabilities and draw a decision curve.
X-tile [26] (version 3.6.1, Yale University School of Medicine, New Haven, Conn) was used to determine the cutoff value of the prognostic score, and patients were divided into high-risk groups and low-risk groups. The difference in survival curves between the high-risk group and the low-risk group was evaluated by a weighted log-rank test (G-rho rank test, rho = 1) [27]. Time-dependent areas under the receiver operating characteristic curve (AUROC) for OS could also be generated by using the multivariable model.
Genetic studies of the radiomics nomogram
In order to explore the biological basis of the prognostic risk score grouping obtained by the radiomics nomogram, it was necessary to conduct related evaluations of potential molecular biology pathways. We used the 4 × 44 K whole human genome expression microarray (Agilent design ID 026,652, GEO accession number GPL13497) to get profiles the expression of 27,958 genes among the 28 pairs of qualified samples. We used GeneSpring GX v12.1 software package to do quantile normalization and subsequent data processing, and gene with low expression or close to the background level was excluded for analyses.
We preformed pre-ranked gene set enrichment analysis (GSEA) approach for genetic analysis as in previously published studies [28,29,30]. Gene set enrichment analysis (GSEA) derives its power by focusing on gene sets, that is, groups of genes that share common biological function, chromosomal location, or regulation [31]. Gene expression values were correlated with the prognostic risk score to rank all genes by using Spearman’s rank correlation coefficient, and we put this gene rank into a ranked gene set enrichment analysis (GSEA) software version 4.2.0. We tested expert-curated pathways from C2 Reactome collection available at MSigDB [32] database cp.kegg.v72.symbols GMT data set. The weighted enrichment analysis method in GSEA was used to conduct enrichment analysis by random combination of 1000 times, and adj. p value was calculated as per to correct for multiple hypothesis testing. The normalized enrichment score (NES) of GSEA software was used to quantify the correlation of the genes with pathways.
The overview of our pipeline is given in Fig. 1.

Flow chart of radiomics. A. Acquisition image data and segment the region of interest for the entire tumor; B. extract multiple quantitative features from the CT image, quantify the shape, first order, texture, Laplacian of Gaussian (LOG) texture and wavelet texture of the tumor-related information. D. After multiple screening, radiomics characteristics were comprehensively analyzed with clinical data and gene expression data
Statistical analysis
The statistical analysis was performed with R software (version 4.0.4, https://www.R-project.org). The packages in R used in this study are described in Supplementary material E3.
In order to evaluate the difference of the clinical information between the training and validation data sets, the Mann–Whitney U test was used for continuous variables (mean survival time) and the classification variables (gender, age, stage, smoking status, degrees of differentiation, CTC count, PD-L1 expression) using χ2 test (Table 1). The levels of statistical significance reported were double-sided, and p value < 0.05 was considered as significant.
Results
Patient clinical characteristics
The clinical characteristics of the training data set and the validation data set are shown in Table 1. A total of 180 patients (102 male and 78 female patients; mean age, 58.39 years ± 7.75; range, 35–81 years) were included in this study (Table 1). The difference was not statistically significant in terms of clinical characteristics including gender, age, smoking status, stage, tumor differentiation, CTC count, PD-L1 expression, Rad-score and OS.
Univariate Cox Proportional Hazards Regression Analysis (Table 2) in the training data set showed that the differences of age, stage, CTC count and Rad-score were statistically significant with p < 0.001.
Radiomics features selection and radiomics score calculation
Selecting all features with high stability (ICC > 0.8) among 180 patients resulted in 1706 radiomics features (28 shape features, 30 first-order intensity statistics features, 62 texture features, 731 LoG features and 855 wavelet features) (Supplementary Fig. 2). And then, the retained features were further screened using the LASSO-Cox method (Supplementary Fig. 3). Finally, 7 radiomics features with nonzero weighting coefficients were obtained (Supplementary Fig. 4), and the corresponding rad-scores were calculated, respectively, (Supplementary Fig. 5). The calculation formula is shown in Supplementary material E4.
There was no statistical difference between the rad-scores in the training set and the validation set (p = 0.735) (Table 1), and it was an independent risk factor for prognosis (hazard ratio (HR) = 3.18) (Table 2).
Radiomics nomogram construction
The clinicopathological nomogram model demonstrated promising clinical prognosis prediction performance (C-index: 0.765; 95% CI: 0.692–0.837). After combining the optimal rad-score with independent clinical prognostic characteristics, the prediction performance of the radiomics nomogram had been significantly improved (C-index: 0.815; 95% CI: 0.756–0.874) (Supplementary Fig. 6). The radiomics nomogram is presented in Fig. 2A. The nomogram calibration curves of the survival probability at 2, 3 and 4 years after diagnosis are shown in Fig. 2B, and they indicate the degree of consistency between the predicted value of the nomogram and the actual observation results.

Comparison of prognostic prediction performance between radiomic nomogram and clinicopathological nomogram. A. The radiomics nomogram constructed using the training data set was used to predict the prognostic risk of patients with “driver gene-negative” LUAD. B. The calibration curves demonstrated that the radiomics nomogram had a good prediction performance of the survival probability at 2, 3 and 4 years after diagnosis. C. The decision curve showed that radiomics nomogram (solid brown line) was a model with a higher net income than clinicopathological nomogram (solid orange line) under most of the given thresholds. D. The integrated discrimination index (IDI) indicated that the prediction performance of radiomics nomogram model was 6.5% higher than that of clinicopathological nomogram model. E. The estimated concordance index (C-index) and Akaike information criteria (AIC) of the radiomic nomogram and clinicopathological nomogram
The decision curve analysis in Fig. 2C demonstrated that across the most reasonable threshold probability range, the overall net benefit of the radiomics nomogram was higher than that of the clinicopathological nomogram. The IDI in Fig. 2D showed that the prediction performance of radiomics nomogram model was 6.5% higher than that of clinicopathological nomogram model. Figure 2E shows the estimated C-index and AIC of the models.
The clinical prognostic risk score of each patient according to the radiomics nomogram was calculated. The optimal cutoff risk point of 65.28 was found out by using X-tile (Supplementary Fig. 7) software and divided the patients into high-risk (> 65.28) and low-risk (≤ 65.28) groups. The prognostic score was significantly correlated with OS in the training data set (p < 0.001, HR = 7.15 (3.93–12.99)), and it was verified in the validation data set (p < 0.001, HR = 8.72 (3.00–25.30)) (Fig. 3). Patients with lower-risk score had better OS. AUROC of training set was 0.88 for 2 years and 0.85 for 4 years. Among validation set, the AUROC was 0.71 for 2 years and 0.78 for 4 years (Fig. 3).


The graph showed the results of the OS curve of Kaplan–Meier survival analysis and time-dependent AUC plot in the training data set (A) and validation data set (B) after dividing the clinical prognosis score into a high-risk group and a low-risk group according to the cutoff value. The dotted line represents the corresponding median survival time
Genetic studies of the radiomics nomogram
We explored the biological basis of the radiomics model through a GSEA analysis based on the RNA expression data of 27 LUAD patients. Available from GSEA results, there were 3 significantly enriched pathways in low-risk score group and 6 in high-risk score group (p < 0.05) (Supplementary Fig. 8). The eight pathways of high-risk score group (primary immunodeficiency, graft versus host disease, antigen processing and presentation, N-glycan biosynthesis, ribosome and hematopoietic cell lineage) and top three pathways of low-risk group (pentose and glucuronate interconversions, O-glycan biosynthesis and drug metabolism other enzymes) are shown in Fig. 4. The pathways suggested the biological basis for the role of the radiomics model may be due to changes in immune and metabolism-related pathways.

GSEA analysis for prediction model most significantly enriched pathways with enrichment scores was shown in different colors. Above the abscissa were pathways enriched in the low-risk score group, below the abscissa were pathways enriched in the high-risk score group
Discussion
In this study, the individualized nomogram based on radiomics characteristics, age, staging and CTC count showed promising prediction efficacy of survival in patients with stage I–III “driver gene-negative” LUAD, with C-index of 0.815 (95% CI: 0.756–0.874). GSEA results showed that the low-risk group was directly related to amino acid metabolism, and the high-risk score group was related to immune and autophagy pathways.
Previous studies had shown that CTCs count was an independent prognostic factor of NSCLC [33], especially in advanced NSCLC [34, 35]. However, there were few studies on the correlation between the prognosis of “driver gene-negative” LUAD after operation and the count of CTCs. Our research confirms that CTC was an independent factor of OS in this genetically unique subset of patients. It can not only predict the prognosis before treatment, but also help monitor the dynamic response of patients during treatment [36] and changes in survival probability [37].
In this study, we used the prognostic nomogram of pre-treatment imaging features and clinical data to stratify the risk of patients with “driver gene-negative” LUAD after surgery. As the first study to use radiomics features in the survival assessment of this genetically unique subset of patients, we found that the combination of radiomics and clinical features has stronger predictive power than a single radiomics feature or clinical feature nomogram, with a higher C-index and better calibration. The decision curve analysis showed that within the most reasonable threshold probability range, the radiomics model performs better than the clinicopathological model. In this study, we focused on patients with “driver gene-negative” LUAD, which was a relatively rare subtype of NSCLC, and mutation-negative patients have limited treatment options and poor prognosis. We developed an imaging–clinical comprehensive prognostic model and conducted preliminary exploration of related pathways in order to guide individual postoperative care for those patients, especially high-risk patients.
The GSEA results showed the low-risk group was directly linked to metabolism-related pathways and the high-risk group correlated with immune-related pathways. Previous studies have also shown that amino metabolism-related pathways [38] play an important role in the development of LUAD. In addition, in this study we found that the high-risk group was related to immune and autophagy pathways, which may indicate that immunotherapy may play a therapeutic role in this population. Thus, these findings provide new treatment orientation for “driver gene-negative” postoperative patients who cannot undergo targeted therapy.
There were also some limitations in our research. First, the inevitable selection bias in patient screening and the small size data set in this study not only greatly influence on the robustness of the radiomics model but also limit the generality of our model. The sample size needs to be expanded to make the results more convincing, and the results require further independent external validation before widespread implementation in clinical practice. Second, despite the promising findings of the present study, the results cannot be generalized to other populations because gene mutation rate and other clinical factors such as smoking can be affected by race. Third, our research mainly focused on patients with LUAD and ignored other histological subtypes of lung cancer patients. Finally, gene test and some of the CTC tests were performed retrospectively; thus, the research results will inevitably be biased due to the preservation duration of the study samples.
Conclusions
The radiomics nomogram was promising to be used as a biomarker for risk stratification for OS in patients with “driver gene-negative” LUAD. The radiomics nomogram well demonstrated the incremental value to the traditional staging system and other clinical–pathological risk factors for individualized OS estimation. Furthermore, the subsequent gene enrichment analysis can not only explain the mechanism of prognosis difference, but more importantly to provide auxiliary evidence for guiding the postoperative treatment plan for this genetically unique subset of patients.
Abbreviations
- AIC:
-
Akaike information criteria
- AUROC:
-
Areas under the receiver operating characteristic curve
- C-index:
-
Concordance index
- CT:
-
Computed tomography
- CTC:
-
Circulating tumor cell
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene set enrichment analysis
- ICC:
-
Intraclass correlation coefficient
- ICIs:
-
Immune checkpoint inhibitors
- IDI:
-
Integrated discrimination improvement
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- LOG:
-
Laplacian of Gaussian
- LUAD:
-
Lung adenocarcinoma
- OS:
-
Overall survival
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed death ligand-1
- Rad-score:
-
Radiomics score
- TPS:
-
Tumor proportion scores
References
Siegel RL, Miller KD, Fuchs HE et al (2021) Cancer statistics, 2021. CA Cancer J Clin 71(1):7–33. https://doi.org/10.3322/caac.21654
Liu Y, Liang L, Ji L et al (2021) Potentiated lung adenocarcinoma (LUAD) cell growth, migration and invasion by lncRNA DARS-AS1 via miR-1885-p/ KLF12 axis. Aging (Albany NY). https://doi.org/10.18632/aging.203632
Cui Y, Fang W, Li C et al (2019) Development and validation of a novel signature to predict overall survival in “driver gene-negative” lung adenocarcinoma (LUAD): results of a multicenter study. Clin Cancer Res 25(5):1546–1556. https://doi.org/10.1158/1078-0432.Ccr-18-2545
Ettinger DS, Wood DE, Aggarwal C et al (2019) NCCN guidelines insights: non-small cell lung cancer, version 1.2020. J Natl Compr Canc Netw 17(12):1464–1472. https://doi.org/10.6004/jnccn.2019.0059
Saito M, Suzuki H, Kono K et al (2018) Treatment of lung adenocarcinoma by molecular-targeted therapy and immunotherapy. Surg Today 48(1):1–8. https://doi.org/10.1007/s00595-017-1497-7
Hamada K, Tian Y, Fujimoto M et al (2021) DNA hypermethylation of the ZNF132 gene participates in the clinicopathological aggressiveness of ’pan-negative’-type lung adenocarcinomas. Carcinogenesis 42(2):169–179. https://doi.org/10.1093/carcin/bgaa115
Sun H, Liu SY, Zhou JY et al (2020) Specific TP53 subtype as biomarker for immune checkpoint inhibitors in lung adenocarcinoma. EBioMedicine 60:102990. https://doi.org/10.1016/j.ebiom.2020.102990
Pathak R, Pharaon RR, Mohanty A et al (2020) Acquired resistance to PD-1/PD-L1 blockade in lung cancer: mechanisms and patterns of failure. Cancers (Basel) 12(12):38–51. https://doi.org/10.3390/cancers12123851
Reck M, Rodríguez-Abreu D, Robinson AG et al (2019) Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer With PD-L1 tumor proportion score of 50% or greater. J Clin Oncol 37(7):537–546. https://doi.org/10.1200/jco.18.00149
Zhou C, Wang Z, Sun Y et al (2022) Sugemalimab versus placebo, in combination with platinuminternational, based chemotherapy, as first-line treatment of metastatic non-small-cell lung cancer (GEMSTONE-302): interim and final analyses of a double-blind, randomised, phase 3 clinical trial. Lancet Oncol 23(2):220–233. https://doi.org/10.1016/S1470-2045(21)00650-1
Aerts H, Velazquez ER, Leijenaar R et al (2014) Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun 5(1):1–9. https://doi.org/10.1038/ncomms5006
Mazzaschi G, Milanese G, Pagano P et al (2020) Dataset on the identification of a prognostic radio-immune signature in surgically resected non small cell lung cancer. Data Brief 31:105781. https://doi.org/10.1016/j.dib.2020.105781
Lin YC, Lin CH, Lu HY et al (2020) Deep learning for fully automated tumor segmentation and extraction of magnetic resonance radiomics features in cervical cancer. Eur Radiol 30(3):1297–1305. https://doi.org/10.1007/s00330-019-06467-3
Vuong D, Tanadini-Lang S, Wu Z et al (2020) Radiomics feature activation maps as a new tool for signature interpretability. Front Oncol 10:578895. https://doi.org/10.3389/fonc.2020.578895
Xie D, Wang TT, Huang SJ et al (2020) Radiomics nomogram for prediction disease-free survival and adjuvant chemotherapy benefits in patients with resected stage I lung adenocarcinoma. Transl Lung Cancer Res 9(4):1112–1123. https://doi.org/10.21037/tlcr-19-577
Ren L, Zhong X, Liu W et al (2021) Clinical significance of a circulating tumor cell-based classifier in stage IB lung adenocarcinoma: a multicenter. Cohort Study Ann Surg. https://doi.org/10.1097/sla.0000000000004780
Griethuysen J, Fedorov A, Parmar C et al (2017) Computational radiomics system to decode the radiographic phenotype. Can Res 77(21):e104–e107. https://doi.org/10.1158/0008-5472.CAN-17-0339
Huang Y, Liu Z, He L et al (2016) Radiomics signature: a potential biomarker for the prediction of disease-free survival in early-stage (I or II) non-small cell lung cancer. Radiology 281(3):947–957. https://doi.org/10.1148/radiol.2016152234
Kothari G, Korte J, Lehrer EJ et al (2021) A systematic review and meta-analysis of the prognostic value of radiomics based models in non-small cell lung cancer treated with curative radiotherapy. Radiother Oncol 155:188–203. https://doi.org/10.1016/j.radonc.2020.10.023
Harrell FE (2015) Regression Modeling Strategies: With Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis. 2015: Regression Modeling Strategies: With Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis
Tibshirani R (1997) The LASSO method for variable selection in the Cox Model. Stat Med 16(4):385–395. https://doi.org/10.1002/(sici)1097-0258(19970228)16:4%3c385::aid-sim380%3e3.0.co;2-3
Geng H, Li S, Guo Y et al (2020) Survival prediction for patients with lung adenocarcinoma: a prognostic risk model based on gene mutations. Cancer Biomark 27(4):525–532. https://doi.org/10.3233/cbm-191204
Xu F, Zhu W, Shen Y et al (2020) Radiomic-based quantitative CT analysis of pure ground-glass nodules to predict the invasiveness of lung adenocarcinoma. Front Oncol 10:872. https://doi.org/10.3389/fonc.2020.00872
Chipman J, Braun D (2017) Simpson’s paradox in the integrated discrimination improvement. Stat Med 36(28):4468–4481. https://doi.org/10.1002/sim.6862
Nix MG, Rowbottom CG, Vivekanandan S et al (2020) Chemoradiotherapy of locally-advanced non-small cell lung cancer: analysis of radiation dose-response, chemotherapy and survival-limiting toxicity effects indicates a low α/β ratio. Radiother Oncol 143:58–65. https://doi.org/10.1016/j.radonc.2019.07.026
Zheng XQ, Huang JF, Lin JL et al (2019) Incidence, prognostic factors, and a nomogram of lung cancer with bone metastasis at initial diagnosis: a population-based study. Transl Lung Cancer Res 8(4):367–379. https://doi.org/10.21037/tlcr.2019.08.16
Alborelli I, Leonards K, Rothschild SI et al (2020) Tumor mutational burden assessed by targeted NGS predicts clinical benefit from immune checkpoint inhibitors in non-small cell lung cancer. J Pathol 250(1):19–29. https://doi.org/10.1002/path.5344
Grossmann P, Gutman DA, Dunn WD Jr et al (2016) Imaging-genomics reveals driving pathways of MRI derived volumetric tumor phenotype features in Glioblastoma. BMC Cancer 16:611. https://doi.org/10.1186/s12885-016-2659-5
Trebeschi S, Drago SG, Birkbak NJ et al (2019) Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann Oncol 30(6):998–1004. https://doi.org/10.1093/annonc/mdz108
Hosny A, Parmar C, Coroller TP et al (2018) Deep learning for lung cancer prognostication: a retrospective multi-cohort radiomics study. PLoS Med 15(11):e1002711. https://doi.org/10.1371/journal.pmed.1002711
Subramanian A, Tamayo P, Mootha VK et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102(43):15545–15550. https://doi.org/10.1073/pnas.0506580102
Liberzon A, Subramanian A, Pinchback R et al (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27(12):1739–1740. https://doi.org/10.1093/bioinformatics/btr260
Lindsay CR, Blackhall FH, Carmel A et al (2019) EPAC-lung: pooled analysis of circulating tumour cells in advanced non-small cell lung cancer. Eur J Cancer 117:60–68. https://doi.org/10.1016/j.ejca.2019.04.019
Markou A, Tzanikou E, Kallergi G et al (2021) Evaluation of monocarboxylate transporter 4 (MCT4) expression and its prognostic significance in circulating tumor cells from patients with early stage non-small-cell lung cancer. Front Cell Dev Biol 9:641978. https://doi.org/10.3389/fcell.2021.641978
Syrigos K, Fiste O, Charpidou A et al (2018) Circulating tumor cells count as a predictor of survival in lung cancer. Crit Rev Oncol Hematol 125:60–68. https://doi.org/10.1016/j.critrevonc.2018.03.004
Gallo M, De Luca A, Frezzetti D et al (2019) The potential of monitoring treatment response in non-small cell lung cancer using circulating tumour cells. Expert Rev Mol Diagn 19(8):683–694. https://doi.org/10.1080/14737159.2019.1640606
Tognela A, Spring KJ, Becker T et al (2015) Predictive and prognostic value of circulating tumor cell detection in lung cancer: a clinician’s perspective. Crit Rev Oncol Hematol 93(2):90–102. https://doi.org/10.1016/j.critrevonc.2014.10.001
Li Y, He CL, Li WX et al (2020) Transcriptome analysis reveals gender-specific differences in overall metabolic response of male and female patients in lung adenocarcinoma. PLoS ONE 15(4):e0230796. https://doi.org/10.1371/journal.pone.0230796
Funding
This work was supported by Grants from the National Natural Science Foundation of China (82102109) and by Grants from Development Center for Medical Science & Technology National Health Commission of the People’s Republic of China (No. WA2020RW10).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This retrospective study was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat-Sen University’s institute (No.[2013]C-084) and conducted in accordance with the Declaration of Helsinki.
Consent for publication
Our study was not a case report and the need for informed consent for publication was waived by the First Affiliated Hospital of Sun Yat-Sen University’s institute due to the non-invasiveness (retrospective analysis of existing data).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, QK., Yang, HS., Shan, SC. et al. A radiomics nomogram prediction for survival of patients with “driver gene-negative” lung adenocarcinomas (LUAD). Radiol med 128, 714–725 (2023). https://doi.org/10.1007/s11547-023-01643-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11547-023-01643-4