
Abstract
Background
Cell-cycle regulators are mutated in approximately 40% of all cancer types and have already been linked to worse outcomes in non-small cell lung cancer adenocarcinomas treated with osimertinib. However, their exact role in osimertinib resistance has not been elucidated.
Objective
In this study, we aimed to evaluate how the CDK4/6-Rb axis may affect the sensitivity to osimertinib.
Methods
We genetically increased the level of CCND1 (Cyclin D1) and reduced the levels of CDKN2A (p16) in two different adenocarcinoma cell lines, PC9 and HCC827. We also retrospectively evaluated the outcome of patients with epidermal growth factor receptor-mutated advanced non-small cell lung cancer depending on their level of Cyclin D1 and p16.
Results
The modified clones showed higher proliferative capacity, modifications in cell-cycle phases, and higher migratory capacity than the parental cells. Cyclin D1-overexpressing clones were highly resistant to acute osimertinib treatment. CDKN2A knockdown conferred intrinsic resistance as well, although a longer time was required for adaption to the drug. In both cases, the resistant phenotype was epidermal growth factor receptor independent and associated with a higher level of Rb phosphorylation, which was unaffected by osimertinib treatment. Blocking the phosphorylation of Rb using abemaciclib, a CDK4/6 inhibitor, exerted an additive effect with osimertinib, increasing sensitivity to this drug and reverting the intrinsic resistant phenotype. In a group of 32 patients with epidermal growth factor receptor-mutated advanced non-small cell lung cancer, assessed for Cyclin D1 and p16 expression, we found that the p16-deleted group presented a lower overall response rate compared with the control group.
Conclusions
We conclude that perturbation in cell-cycle regulators leads to intrinsic osimertinib resistance and worse patient outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Overexpression of CCND1 or knockdown of CDKN2A leads to osimertinib resistance. |
The short time for the onset points to an intrinsic resistance. |
Resistance is epidermal growth factor receptor independent and associated with Rb hyper-phosphorylation. |
Abemaciclib exerts an additive effect with osimertinib and reverts resistance. |
1 Introduction
Non-small cell lung cancer (NSCLC) is the most common type of lung cancer and the leading cause of cancer-related mortality worldwide. Mutations in the epidermal growth factor receptor (EGFR) are found in 40–60% of South-East Asian patients and 10–20% of Caucasian patients with NSCLC [1] and are associated with high sensitivity to EGFR-tyrosine kinase inhibitors (EGFR-TKIs). Osimertinib is a third-generation TKI originally designed to target EGFR-sensitizing mutations and the T790M mutation, which represents the most common acquired resistance mechanism to first- and second-generation EGFR-TKIs [2]. In 2018, osimertinib was approved as a first-line treatment for EGFR-mutated NSCLC following FLAURA trial results, which demonstrated the advantage of osimertinib in terms of overall survival (OS) and progression-free survival (PFS) [3, 4] compared with previous-generation EGFR-TKIs. However, as with previous EGFR-TKIs, resistance to osimertinib eventually occurs [5, 6], and the described mechanisms are highly heterogeneous, including EGFR-dependent or EGFR-independent mechanisms [7, 8]. Moreover, a fraction of patients with EGFR-mutated NSCLC is intrinsically insensitive to osimertinib and clinically non-responders, but the underlying mechanisms are still unknown [8, 9].
Cell-cycle regulation depends on the activity of cyclin dependent kinases (CDKs), which in response to mitogenic signals, bind to cyclin proteins, induce hyper-phosphorylation of Rb protein, and promote the release of E2F transcription factor with subsequent progression of the cell cycle through the S phase. CDK4/6 activity is regulated by a family of proteins, including p16, which inhibits the activity of the CDK4/6-Cyclin D1 complex, preventing the phosphorylation of Rb and the release of E2F, thus blocking the cell cycle at the G1 phase.
Dysregulation of the CDK4/6-Cyclin D-Rb-E2F pathway due to genetic alterations is a common feature of many human cancers and can account for more than 40% of patients with cancer [10]. Amplification of Cyclin D1 gene (CCND1) or Cyclin D1 overexpression, CDK4 gene amplification, and inactivation of p16 through CDKN2A deletion, methylation, or mutation has been reported in several solid tumors including NSCLC [11,12,13,14].
Cell-cycle regulators have been also investigated for their role in resistance to cancer therapy [15]. Alterations in genes related to the cell cycle have been detected in 10–12% of patients who progressed on EGFR-TKIs [16, 17]; in particular, the master regulator Rb, coded by the RB1 gene, has been identified as a key player in osimertinib resistance [8, 17, 18].
Therefore, it is important to understand the potential role of dysregulation in cell-cycle regulators in resistance to osimertinib, particularly because drugs targeting the Rb pathway are now available and show promising activity against multiple cancers [19]. Three CDK4/6 inhibitors, including abemaciclib (LY835219), are currently approved for the treatment of hormone receptor-positive, human epidermal growth factor receptor 2-negative, metastatic breast cancer [20]. By inhibiting CDK4/6 proteins and blocking the transition from G1 to S phase of the cell cycle, CDK4/6 inhibitors can block cell-cycle progression and prevent cancer cell proliferation. Our previous studies [18, 21,22,23] elucidated the efficacy of abemaciclib in NSCLC, malignant pleural mesothelioma, breast cancer, and hepatocarcinoma cells by studying its effects on cell survival, senescence, and cell metabolism.
In the current study, we investigated the impact of Cyclin D1 amplification or p16 downregulation on osimertinib resistance in EGFR-mutated NSCLC cells. We also explored the potential of combining osimertinib with abemaciclib to restore sensitivity in PC9 and HCC827 cells modified to carry alterations in CDKN2A or CCND1 genes, and retrospectively evaluated the outcome of patients with EGFR-mutated advanced NSCLC in relation to levels of Cyclin D1 and p16.
2 Materials and Methods
2.1 Cell Lines and Culture
The PC9 cell line was provided by Dr. P. Jänne (Dana-Farber Cancer Institute). The HCC827 cell line was purchased from ATCC (Manassas, VA, USA). Both PC9 and HCC827 cell lines have an exon 19 deletion in the EGFR gene. Cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) [Invitrogen, Carlsbad, CA, USA] and PenStrep (Invitrogen) at 37 °C and 5% CO2. Osimertinib (AstraZeneca, Milan, Italy) and abemaciclib (Selleckchem, Houston, TX, USA) were dissolved in DMSO (Sigma Aldrich, St Louis, MO, USA) and administered to cells at a final concentration lower than 0.1% DMSO.
2.2 Viral Infection
To modify the cell genome, cells were plated at 60% confluence. The next day, cells were treated with Viral Lentiparticles (Origene, Rockville, MD, USA) carrying the desired open reading frame (for CCND1 overexpression) or a mix of four different short hairpin RNAs targeting different parts of the CDKN2A gene (for CDKN2A knock-down) and polybrene 5 µg/mL in a medium without antibiotics. After 24 h, the medium was replaced with a fresh medium. After 72 h, the cells were selected in puromycin 1 µg/mL for 2 weeks. The clones were then handpicked and cultured for analysis and characterization.
2.3 Analysis of Cell Proliferation and Cell Cycle
Cells were plated in 96-well plates (5000 cells per well) for dose–response experiments. Cell proliferation was evaluated using a crystal violet (CV) assay, as previously described [18]. Briefly, the cells were fixed in ice-cold methanol at 4 °C for 15 minutes and stained with 0.1% CV (Sigma-Aldrich) for 15 minutes. The dye was washed with distilled water. Crystal violet was solubilized with 0.5% Triton × 100 in phosphate buffered saline. The absorbance was measured at 570 nm. The distribution of the cells in the cell-cycle phases was determined by propidium iodide staining and a subsequent analysis using a CytoFLEX Flow Cytometer (Beckman Coulter Life Sciences, Indianapolis, IN, USA), as described elsewhere [24].
2.4 Colony Formation Assay
For colony formation experiments, 500 cells per well were plated in a six-well plate. The next day, cells were treated with the desired concentration of drugs, and the medium with drugs was replaced every 3 days. At the end of the experiment, the cells were stained with CV. Crystal violet was then solubilized with 0.5% Triton × 100 in phosphate buffered saline. The absorbance was measured at 570 nm.
2.5 Cell Migration Assay
Cell migration analyses were performed as previously described [25]. Briefly, 1 × 105 cells were plated in a 6.5-mm Transwell® with an 8.0-µm Pore Polycarbonate Membrane Insert (Corning, NY, USA) in a 24-well plate. After 16 h, the cells in the upper chamber that did not migrate were removed using a cotton swab. The inserts were then fixed and stained with methanol and CV. Filters from the Transwell were cut and fixed on microscope slides. Images were taken at 20× on a phase contrast microscope, and the cells were counted.
2.6 Real-Time Polymerase Chain Reaction
RNA was extracted using a Total RNA Extraction KIT (RBC Bioscience, New Taipei City, Taiwan), according to the manufacturer’s instructions. Next, 500 ng of RNA was reverse transcribed into cDNA with a Primescript RT Mastermix (Takara Bio Europe SAS, Saint-Germain-en-Laye, France) and quantified in a StepOne machine (Applied Biosystems, Waltham, MA, USA) using TB Green Premix Ex Taq II (Takara).
2.7 Western Blot Analysis
Proteins were extracted and analyzed as described in [26]. Antibodies against phospho-Rbser807/ser811, Rb, Cyclin D1, p16INK4a, phospho-EGFRtyr1068, EGFR, phospho- ERK1/2, ERK1/2, and actin, and HRP-conjugated secondary antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA), and the chemiluminescence system (ImmobilionTM Western Chemiluminescent HRP Substrate) was from Millipore (Temecula, CA, USA). The reagents for electrophoresis and blotting analysis were obtained from Bio-Rad (Hercules, CA, USA).
2.8 Immunohistochemistry Analysis
Immunohistochemical staining of formalin-fixed paraffin-embedded tissue sections was performed on all samples. After deparaffinization and rehydration, sections were treated with 3% hydrogen peroxidase for 5 minutes. For antigen retrieval, sections were treated with pH9 Tris-EDTA buffer for 30 minutes in a water bath at 98 °C, followed by staining with the following primary antibodies: Ventana Anti-Cyclin D1 (clone SP4-R; Roche, Basel, Switzerland) and p16 (clone E6H4; Roche). The sections were immunostained using a polymeric system Ultraview DAB Detection Kit (Ventana-Roche, Basel, Switzerland) in accordance with the manufacturer’s specifications.
Diaminobenzidine was used for staining, and the sections were counterstained with hematoxylin. Negative controls consisted of substituting normal serum for the primary antibody. The percentage of Cyclin D1-positive cells (nuclear staining) was calculated as follows at 20×. Data were analyzed as a continuous variable and transformed into a categorical binary variable (positive vs negative; qualitative measurement) using a cut-off value of at least 1% positive cells to define a case as positive. The percentage of p16-positive cells (strong nuclear and cytoplasmic expression) was calculated in a continuous segment of cells (at least 10–20 cells) at 20×.
Overall response rate was defined as the proportion of patients who achieved a complete response or partial response as their best overall response at the radiological evaluation measured according to the standard RECIST criteria version 1.1. Progression-free survival was defined as the time from osimertinib initiation to objective disease progression or death due to any cause. Overall survival was defined as the time from osimertinib initiation to the date of death, regardless of cause.
2.9 Statistical Analysis
Statistical analyses were performed using the GraphPad Prism version 9.0 software (GraphPad Software Inc., San Diego, CA, USA). Results are expressed as mean ± standard deviation (SD). Differences between the mean values recorded for different experimental conditions were evaluated by the Student’s t-test or one-way analysis of variance followed by Bonferroni’s post-test, and p-values are indicated where appropriate in the figures and in their legends. The median PFS and OS were calculated using the Kaplan–Meier method. Statistical differences were assessed using the log-rank test. Adjusted p-values of less than 0.05 were considered significant.
3 Results
3.1 Perturbation in the p16-Cyclin D1 Axis Leads to Higher Proliferative and Migratory Capacity
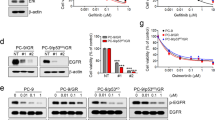
To understand the potential role of cell-cycle regulators in resistance to osimertinib, we increased the expression of Cyclin D1 or reduced the p16 level in two EGFR-mutated NSCLC cell lines, PC9 and HCC827. This was achieved using lentiviral particles carrying the open reading frame for CCND1 (Cyclin D1) or four different short hairpin RNAs for CDKN2A (p16). To confirm the efficacy of both overexpression and knockdown (Fig. 1A), we selected one subclone for each genetic alteration and for each cell line (#6 for PC9 p16-, #3 for PC9 CycD1+, #3 for HCC827 p16-, and #1 for HCC827 CycD1+). Once the expression of the gene alteration of interest was confirmed by real-time polymerase chain reaction quantification (Fig. 1B), we studied the biological effects of these alterations to unveil the role of cell-cycle regulators in osimertinib resistance.

p16 Knock-down and Cyclin D1 overexpression efficiency. A Proteins were extracted from different clones and from control cells (NC, transfected with non-coding RNA), run on polyacrylamide gels and blot for Cyclin D1 and p16 to assess the efficacy of the transfection procedure. B Clones for each genetic modification and for each cell line (#6 PC9p16-; #4 PC9 CycD1+; #3 HCC827 p16-; #1 HCC827 CycD1+) were selected and messenger RNA was assessed through real-time polymerase chain reaction (non-coding cells relative quantification = 1). Results are means ± standard deviations of three independent experiments
We evaluated the effects of Cyclin D1 overexpression (CycD1+) or p16 reduction (p16-) on the cell growth rate by a CV assay in the two different models. As shown in Fig. 2A, both Cyclin D1-overexpressing cells and CDKN2A knockdown cells exhibited a higher proliferative capacity than control cells. As expected, the distribution of cells in the different phases of the cell cycle, evaluated by flow cytometry, was different in the clones compared with the respective control cells. As shown in Fig. 2B, p16- and CycD1+ cells were characterized by a larger proportion of cells in the S phase and a lower percentage of cells in the G0/G1 phase. Both p16- and CycD1+ clones displayed increased activation of Rb protein in the presence of a reduced level of FBS, confirming that the genetic modifications lead to higher downstream activation of the Rb pathway (Fig. 2C).

Dysregulation of cell-cycle regulators leads to higher proliferative and migratory capacity. A Cells were seeded in a 96-well plate and cell growth was evaluated by a crystal violet assay. Results are expressed as fold increase over T0 and are means ± standard deviations of three independent experiments. B The indicated cells were stained with propidium iodide and their distribution in cell-cycle phases was determined by flow cytometry. Results are means ± standard deviations of at least two independent experiments. C The different clones were cultured in a complete growth medium (first lane) or in a medium deprived of FBS. After 48 h in one condition (2° lane), the medium was changed from 0.5% to 10% of FBS. Then, after 6 h, proteins were extracted and analyzed by western blotting for the indicated proteins. D Cell migration was evaluated as indicated in the Materials and Methods section. Results are from three independent experiments. * p < 0.05; **p < 0.01; ***p < 0.001 vs NC
Finally, we investigated the effects of Cyclin D1 overexpression and p16 deletion on cell migratory capacity. The results shown in Fig. 2D indicate that both genetic alterations led to higher migration capacity in PC9- and HCC827-mutated cell clones than in control cells, suggesting a more aggressive phenotype, as previously reported [27].
3.2 Cyclin D1 Overexpression or p16 Deletion Leads to EGFR-Independent Intrinsic Resistance to Osimertinib
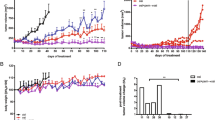
The main purpose of this study was to understand the relationship between cell-cycle regulator modifications and osimertinib resistance. To evaluate the differences in the response to osimertinib between the different clones and the corresponding parental cells, we treated the cells with increasing concentrations of the drug for 72 h. As shown in Fig. 3A, p16- clones did not show a different sensitivity to osimertinib compared to control cells infected with non-coding RNA (IC50 PC9 non-coding = 2.36 nM ± 1; PC9 p16- = 1.78 ± 0.5 nM; HCC827 = 5.8 ± 3 nM; HCC827 p16- = 1.3 ± 2 nM), whereas CycD1+ clones showed higher resistance to the drug (IC50 PC9 CycD1+ = 512 ± 35 nM; HCC827 CycD1+ = 604 ± 43 µM). Interestingly, in contrast to parental cells, osimertinib treatment did not significantly perturb the distribution of the cells in the cell-cycle phases, not only in CycD1+ clones but also in p16- clones (Fig. 3B).

Cyclin D1 overexpression or p16 deletion leads to intrinsic resistance to osimertinib. A The indicated cell lines were treated with increasing concentrations of osimertinib for 72 h. Cell proliferation was assessed by a crystal violet assay; for each cell model, the data are expressed as percentage versus the corresponding untreated control cells and are means ± standard deviations of three independent experiments. B The indicated cells were treated with osimertinib 100 nM for 24 h and then stained with propidium iodide, and their distribution in cell-cycle phases was determined by flow cytometry. Results are means ± standard deviations of at least two independent experiments. C Cells from the indicated clones were treated with osimertinib 200 nM for 14 days (the medium was replaced every 72 h). Then, cell proliferation was assessed by a crystal violet assay and images were taken. Results are means ± standard deviations of at least two independent experiments. D Cells from the indicated clones were treated with escalating dose of osimertinib (20 nM, 50 nM, 100 nM, 200 nM) for 1 week (medium with drug was replaced every 72 h). Finally, cell proliferation was assessed by a crystal violet assay and images were taken. Results are means ± standard deviations of at least two independent experiments. E The different clones were treated with osimertinib 100 nM for 24 h and the level of the indicated proteins was assessed through western blotting. *p < 0.05; **p < 0.01; ***p < 0.001 vs NC
One limitation of assessing drug resistance through short-term dose–response experiments is that it highlights the ability to overcome the cytotoxic effect of a compound rather than showing the ability of the cells to grow in the presence of the drug. Therefore, to better elucidate the effect of osimertinib on cells carrying genetic alterations in cell-cycle regulators, we shifted from short- to long-term experiments. For a colony formation assay, we seeded all cell lines at low confluence (500 cells in six-well plates) and treated them with 200 nM (10× IC50) osimertinib for 14 days (Fig. 3C). As expected, control cells did not grow at this concentration. In contrast, CycD1+ and p16- clones were able to proliferate, although to different extents, suggesting that they were intrinsically resistant to osimertinib, as also supported by the cell-cycle analysis. Considering the lower ability of p16- clones to form colonies in the presence of osimertinib compared with CycD1+ clones, we shifted from a single high-dose setting to a dose-escalating setting to better clarify whether the p16- clones were truly resistant. Hence, we treated p16- cells with increasing osimertinib concentrations (range, 20–200 nM), and a high number of colonies were found in both PC9 -and HCC827-derived p16- clones at the end of a 4-week treatment (Fig. 3D). These results confirm that both genetic alterations lead to an intrinsically resistant phenotype to osimertinib.
The western blot analysis demonstrated that EGFR phosphorylation was inhibited by osimertinib in both CycD1+ and p16- clones, indicating that the resistant phenotype was due to EGFR-independent mechanisms. ERK1/2, an important downstream effector of EGFR, was also inhibited by osimertinib treatment, indicating a complete shutdown of this pathway (Fig. 3E).
We then evaluated the effect of osimertinib on the downstream effector Rb. As shown in Fig. 3E, osimertinib completely inhibited Rb phosphorylation in PC9 and HCC827 control cells, as previously reported [18], whereas it had no or little effect on CycD1+ and p16- clones, indicating that phosphorylation of Rb represents the player of resistance in cells with alterations in cell-cycle regulators. Total Rb was also decreased in control cells treated with osimertinib, as previously reported by our group [18], to a less extent in p16- clones but was unaffected in CycD1+ clones.
3.3 Abemaciclib Exerts Antiproliferative Effects in Cyclin D1-Overexpressing and p16 Deleted Clones
If dysregulation of the p16-CDK4/6-Rb axis is responsible for the osimertinib-resistant phenotype, it should be possible to reverse this effect by blocking one of the components of the signaling cascade. Therefore, we tested the efficacy of the CDK4/6 inhibitor abemaciclib in the cell clones, combining a fixed dose (500 nM) of this drug with increasing concentrations of osimertinib. As shown in Fig. 4A, a short-term combined treatment inhibited cell proliferation more strongly than osimertinib alone in both PC9 and HCC827 Cyclin D1-overexpressing cells. To understand the nature of the interaction between abemaciclib and osimertinib, we combined increasing concentrations of both drugs and plotted the results using the SynergyFinder 3.0. The results shown in Fig. 4B indicate that the effects of the drug combination are additive.

Combined treatment with osimertinib and abemaciclib exerts an additive effect. A The indicated clones were treated with increasing concentrations of osimertinib for 72 h and a fixed dose of abemaciclib (500 nM). Cell proliferation was assessed by a crystal violet assay; for each cell model, the data are expressed as a percentage versus the corresponding untreated control cells and are means ± standard deviations of three independent experiments. B The indicated cells were treated with increasing concentrations of osimertinib and abemaciclib. Cell proliferation was assessed by a crystal violet assay and the nature of interaction was analyzed using Synergy Finder 3.0. C Cells from the indicated clones were treated with osimertinib (200 nM) and abemaciclib (500 nM) for 14 days (medium was replaced every 72 h). Then, cell proliferation was assessed by a crystal violet assay and pictures were taken. Results are means ± standard deviations of at least two independent experiments. D Cells from indicated clones were treated with escalating doses of osimertinib (20 nM, 50 nM, 100 nM, and 200 nM for 1 week each, the medium was replaced every 72 h) and a fixed dose of abemaciclib (500 nM). Then, cell proliferation was assessed by a crystal violet assay and pictures were taken. Results are means ± standard deviations of at least two independent experiments. *** p < 0.001 vs untreated cells
Then, we evaluated the ability of the cell clones to form colonies after long-term treatments. Figure 4C shows that the drug combination significantly inhibited colony formation in Cyclin D1-overexpressing clones. To appreciate the effects of the drug combination even on p16- clones, we shifted from a high single-dose setting to a dose-escalation setting. The results shown in Fig. 4D demonstrate that the addition of abemaciclib to osimertinib effectively inhibited the ability of p16- clones to form colonies.
As expected, at the molecular level, treatment with abemaciclib inhibited Rb expression and phosphorylation (Fig. 5A), as already reported in several tumor types [22, 28, 29], suggesting that this protein plays a key role in osimertinib resistance. Because of this molecular alteration, abemaciclib treatment increased the fraction of cells in the G0/G1 phase and decreased the fraction of cells in the S-G2/M phase in both CycD1+ and p16- clones (Fig. 5B).

Combined treatment with osimertinib and abemaciclib influences cell proliferation through the Rb pathway. A The indicated cells were treated with abemaciclib 500 nM for 24 h. Proteins were then extracted and analyzed by western blotting for pRb and Rb expression. B The indicated cells were treated with abemaciclib (500 nM) or the combination of abemaciclib (500 nM) and osimertinib (100 nM) for 24 h and then stained with propidium iodide and their distribution in cell-cycle phases was determined by flow cytometry. Results are means ± standard deviations of three independent experiments. *p < 0.05; **p < 0.01 vs untreated cells
3.4 Patients with p16 Deletion Show a Lower Response Rate to Osimertinib
In order to explore the role of Cyclin D1 and p16 alterations in osimertinib resistance, we retrospectively reviewed osimertinib outcomes in a group of 32 patients with advanced EGFR-mutated NSCLC treated with first-line osimertinib who were assessed for Cyclin D1 and p16 immunohistochemistry. Overall, 28 (85.5%) patients had a positive immunohistochemical staining for Cyclin D1, whereas four (12.5%) patients were Cyclin D1 negative. Among Cyclin D1-positive patients, p16- patients had a significantly lower overall response rate compared with p16+ patients (61% vs 100%, respectively, p = 0.030). No statistically significant differences in median PFS and OS were observed between the two groups.
4 Discussion and Conclusions
Here, we show that modifications in the expression of cell-cycle regulators can lead to intrinsic osimertinib resistance. Combination treatment with osimertinib and abemaciclib has already been proposed to overcome acquired resistance in resistant cell lines by our group [18, 21, 22].
The possibility of targeting cell-cycle alterations is not new in oncology research because they represent one of the most dysregulated pathways, with over 40% of cancers presenting a mutation in CDKs or cyclins [10]. Alterations in RB1 have been observed in EGFR-mutant NSCLCs that have transformed to small cell lung carcinoma [30] and are associated with worse outcomes and osimertinib resistance [31, 32]. Mutations in cell-cycle regulators are also found in more than 10% of patients who present resistance to first- or second-line osimertinib treatment [8, 30, 31, 33]. In particular, a homozygous p16/CDKN2A deletion has been correlated with a lower response rate (48% vs 78%) and shorter PFS (5.3 months vs 10.5 months), pointing to the critical role of cell-cycle regulators in patient outcomes and drug responses [33].
In our previous study, we investigated the role of phosphorylated Rb as a biomarker and target in osimertinib-resistant NSCLC cells and demonstrated that Rb phosphorylation was maintained in NSCLC cell lines with different mechanisms of acquired resistance to osimertinib [18]. This study aimed to elucidate the role of the p16-CDK-Rb pathway in osimertinib resistance. Here, we demonstrate that Cyclin D1 overexpression and p16 knockdown lead to intrinsic osimertinib resistance. However, even if both the genetic modifications have the same effect on increasing the phosphorylation of Rb, they unexpectedly produce different responses to osimertinib. Cyclin D1-overexpressing clones show a strong resistance to short-term treatment, while p16 deleted cells need a longer time period and long-term treatment to display the resistant phenotype. However, it is important to highlight the different approaches and biological principles behind the two modifications. Overexpressing an oncogene is different from knocking down an oncosuppressor that relies on a highly redundant pathway. It is likely that the effect in the second case is not as strong as in the first case. This hypothesis is supported by the fact that osimertinib treatment did not affect Rb phosphorylation in CycD1+ clones but slightly reduced phosphorylation in p16- clones. The fact that both CycD1+ and p16- clones presented a higher Rb phosphorylation could be an important indication that targeting this pathway can reverse the resistant phenotype.
It is well known that abemaciclib alone can slow down the cell cycle, induce senescence, and act as a cytostatic [34]. We have previously provided preclinical evidence for abemaciclib use to overcome resistance in patients progressing to first-line osimertinib and suggested it as a potential approach to prevent or delay osimertinib resistance in the first-line setting [18]. However, none of the examined cell lines harbored mutations in the cell-cycle regulators. In this study, we evaluated the role of abemaciclib in reverting the resistant phenotype caused by specific genetic alterations in the cell-cycle proteins. We demonstrated that abemaciclib inhibited cell growth in both CycD1+ and p16- clones (in fixed-dose and dose-escalation experiments, respectively) when used in combination with osimertinib. Our data support a clinical trial evaluation of abemaciclib in combination with osimertinib as a first-line treatment for patients presenting with a cell-cycle regulator alteration.
The obvious weakness of targeted therapy, and in general, of all single-drug therapies, is the development of resistance. Combining different compounds with the ability to inhibit different pathways has always been considered a promising therapeutic strategy. The FLAURA2 (NCT04035486) trial is currently exploring the combination of osimertinib and chemotherapy in treatment-naïve patients, and this strategy has already provided encouraging results in preclinical research [35]. Moreover, a combination of MEK inhibitors [36, 37], AXL inhibitors [38], and bevacizumab [39] has been proposed.
To further support the role of cell-cycle regulators in clinical outcomes, we explored first-line osimertinib outcomes in a group of patients with advanced EGFR-mutated NSCLC depending on their immunohistochemical expression of Cyclin D1 and p16. We found that, among Cyclin D1-positive patients, the p16- group was enriched in non-responders compared with the p16+ group, strengthening the key role of cell-cycle regulators in mediating osimertinib resistance. In this context, we can speculate that a combined treatment with CDK inhibitors could be beneficial for this peculiar group of patients. However, because of the small sample size of the Cyclin D1- group, we could not perform additional analyses. Finally, our findings suggest that abemaciclib combined with osimertinib may be a valuable option for those patients with EGFR-mutated NSCLC who have alterations in the p16-Cyclin D1-CDK4/6 pathway.
References
Hsu WH, Yang JC, Mok TS, Loong HH. Overview of current systemic management of EGFR-mutant NSCLC. Ann Oncol. 2018;29:i3-9.
Nagasaka M, Zhu VW, Lim SM, Greco M, Wu F, Ou SI. Beyond osimertinib: the development of third-generation EGFR tyrosine kinase inhibitors for advanced EGFR+ NSCLC. J Thorac Oncol. 2021;16:740–63.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41–50.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378:113–25.
Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv192–237.
Hendriks LE, Kerr KM, Menis J, Mok TS, Nestle U, Passaro A, et al. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34:339–57.
Akli A, Girard N, Fallet V, Rousseau-Bussac G, Gounant V, Friard S, et al. Histomolecular resistance mechanisms to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer: a multicentric retrospective French study. Target Oncol. 2022;17:675–82.
Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121:725–37.
Chamorro DF, Cardona AF, Rodriguez J, Ruiz-Patino A, Arrieta O, Moreno-Perez DA, et al. Genomic landscape of primary resistance to osimertinib among Hispanic patients with EGFR-mutant non-small cell lung cancer (NSCLC): results of an observational longitudinal cohort study. Target Oncol. 2023;18:425–40.
Jhaveri K, Burris Rd HA, Yap TA, Hamilton E, Rugo HS, Goldman JW, et al. The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert Rev Anticancer Ther. 2021;21:1105–24.
Ghafouri-Fard S, Khoshbakht T, Hussen BM, Dong P, Gassler N, Taheri M, et al. A review on the role of cyclin dependent kinases in cancers. Cancer Cell Int. 2022;22:325.
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72.
Tchakarska G, Sola B. The double dealing of cyclin D1. Cell Cycle. 2020;19:163–78.
Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine. 2016;8:30–9.
Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;38:255–71.
Gini B, Thomas N, Blakely CM. Impact of concurrent genomic alterations in epidermal growth factor receptor (EGFR)-mutated lung cancer. J Thorac Dis. 2020;12:2883–95.
Gomatou G, Syrigos N, Kotteas E. Osimertinib resistance: molecular mechanisms and emerging treatment options. Cancers (Basel). 2023;15:841.
La Monica S, Fumarola C, Cretella D, Bonelli M, Minari R, Cavazzoni A, et al. Efficacy of the CDK4/6 dual inhibitor abemaciclib in EGFR-mutated NSCLC cell lines with different resistance mechanisms to osimertinib. Cancers (Basel). 2020;13:6.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
Tolaney SM, Beeram M, Beck JT, Conlin A, Dees EC, Puhalla SL, et al. Abemaciclib in combination with endocrine therapy for patients with hormone receptor-positive, HER2-negative metastatic breast cancer: a phase 1b study. Front Oncol. 2021;11: 810023.
Terenziani R, Galetti M, La Monica S, Fumarola C, Zoppi S, Alfieri R, et al. CDK4/6 inhibition enhances the efficacy of standard chemotherapy treatment in malignant pleural mesothelioma cells. Cancers (Basel). 2022;14:5925.
Digiacomo G, Fumarola C, La Monica S, Bonelli M, Cavazzoni A, Galetti M, et al. CDK4/6 inhibitors improve the anti-tumor efficacy of lenvatinib in hepatocarcinoma cells. Front Oncol. 2022;12: 942341.
Cretella D, Fumarola C, Bonelli M, Alfieri R, La Monica S, Digiacomo G, et al. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci Rep. 2019;9:13014.
Fumarola C, La Monica S, Alfieri RR, Borra E, Guidotti GG. Cell size reduction induced by inhibition of the mTOR/S6K-signaling pathway protects Jurkat cells from apoptosis. Cell Death Differ. 2005;12:1344–57.
La Monica S, Caffarra C, Saccani F, Galvani E, Galetti M, Fumarola C, et al. Gefitinib inhibits invasive phenotype and epithelial-mesenchymal transition in drug-resistant NSCLC cells with MET amplification. PLoS ONE. 2013;8: e78656.
Chou CW, Lin CH, Hsiao TH, Lo CC, Hsieh CY, Huang CC. et al. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci Rep. 2019;9:20403.
Fuste NP, Ferrezuelo F, Gari E. Cyclin D1 promotes tumor cell invasion and metastasis by cytoplasmic mechanisms. Mol Cell Oncol. 2016;3: e1203471.
Guarducci C, Bonechi M, Benelli M, Biagioni C, Boccalini G, Romagnoli D, et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer. 2018;4:38.
Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 2011;17:1591–602.
Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377.
Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49:1693–704.
Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. 2018;24:6195–203.
Jiang J, Gu Y, Liu J, Wu R, Fu L, Zhao J, et al. Coexistence of p16/CDKN2A homozygous deletions and activating EGFR mutations in lung adenocarcinoma patients signifies a poor response to EGFR-TKIs. Lung Cancer. 2016;102:101–7.
Hino H, Iriyama N, Kokuba H, Kazama H, Moriya S, Takano N, et al. Abemaciclib induces atypical cell death in cancer cells characterized by formation of cytoplasmic vacuoles derived from lysosomes. Cancer Sci. 2020;111:2132–45.
La Monica S, Minari R, Cretella D, Flammini L, Fumarola C, Bonelli M, et al. Third generation EGFR inhibitor osimertinib combined with pemetrexed or cisplatin exerts long-lasting anti-tumor effect in EGFR-mutated pre-clinical models of NSCLC. J Exp Clin Cancer Res. 2019;38:222.
Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75:2489–500.
La Monica S, Minari R, Cretella D, Bonelli M, Fumarola C, Cavazzoni A, et al. Acquired BRAF G469A mutation as a resistance mechanism to first-line osimertinib treatment in NSCLC cell lines harboring an EGFR exon 19 deletion. Target Oncol. 2019;14:619–26.
Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O’Brien JP, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene. 2019;38:656–70.
Yu HA, Schoenfeld AJ, Makhnin A, Kim R, Rizvi H, Tsui D, et al. Effect of osimertinib and bevacizumab on progression-free survival for patients with metastatic EGFR-mutant lung cancers: a phase 1/2 single-group open-label trial. JAMA Oncol. 2020;6:1048–54.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Open access funding provided by Università degli Studi di Parma within the CRUI-CARE Agreement. This work was supported by the Italian Association for Cancer Research, Milan (grant IG2017-20074 PI M. Tiseo), by AstraZeneca, Milan Italy, and by the program “FIL-Quota Incentivante” of the University of Parma and co-sponsored by Fondazione Cariparma. The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Ethical approval and consent to participate
The study was approved by the institutional review board of University-Hospital of Parma (Protocol 14033, 09/04/2018) and informed consent was obtained from all individual participants included in the study. All procedures performed in studies involving human participants were in accordance with the Helsinki declaration as revised in 2013 and its later amendments.
Conflict of interest
FV, SLM, AL, LG, MB, AC, CF, MG, KE, RM, PGP, MT, and RA have no conflicts of interest that are directly relevant to the content of this article. MT received speaker and consultant fees from AstraZeneca, Pfizer, Eli-Lilly, BMS, Novartis, Roche, MSD, Boehringer Ingelheim, Otsuka, Takeda, and Pierre Fabre. MT has received institutional research grants from AstraZeneca and Boehringer Ingelheim. RA has received institutional research grants from AstraZeneca.
Consent for Publication
Not applicable.
Availability of data and material
All data generated or analyzed during this study are included in the article.
Code availability
Not applicable.
Author contributions
Conceptualization: FV, SLM, PGP, MT, RA; data curation: MB, MG, CF; formal analysis: AC, AL, RM; funding acquisition: MT, RA; investigation: FV, SLM, LG, KE; supervision: PGP, RA, MT; writing, original draft: FV, SLM; writing, review and editing: PGP, MT, RA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Volta, F., La Monica, S., Leonetti, A. et al. Intrinsic Resistance to Osimertinib in EGFR Mutated NSCLC Cell Lines Induced by Alteration in Cell-Cycle Regulators. Targ Oncol 18, 953–964 (2023). https://doi.org/10.1007/s11523-023-01005-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-023-01005-0