
Abstract
Introduction
Fibroblast growth factor receptor (FGFR)-4/FGF19 pathway dysregulation is implicated in hepatobiliary and other solid tumors. INCB062079, an oral, selective, FGFR4 inhibitor, inhibits growth in FGF19/FGFR4-driven liver cancer models.
Methods
This was a two-part, phase I study (NCT03144661) in previously treated patients with advanced solid tumors. The primary objective was to determine safety, tolerability, and maximum tolerated dose (MTD), while secondary objectives included pharmacokinetics, pharmacodynamics (plasma FGF19; bile acid salts/7α-hydroxy-4-cholesten-3-one [C4] levels), and preliminary efficacy. In Part 1, patients received INCB062079 starting at 10 mg once daily, with 3 + 3 dose escalation. Part 2 (dose expansion) was not conducted because of study termination.
Results
Twenty-three patients were treated (hepatobiliary, n = 11; ovarian, n = 9; other, n = 3). Among six patients receiving 15 mg twice daily, two patients had dose-limiting toxicities (DLTs; grade 3 diarrhea, grade 3 transaminitis). Both had high pretreatment C4 concentrations, prompting a protocol amendment requiring pretreatment C4 concentrations < 40.9 ng/mL and concomitant prophylactic bile acid sequestrant treatment. No additional DLTs were reported at 10 and 15 mg twice daily; higher doses were not assessed. The most common toxicity was diarrhea (60.9%). INCB062079 exposure was dose-proportional; FGF19 and bile acid/C4 concentrations increased with exposure. One partial response was achieved (15 mg twice daily; ovarian cancer; FGF/FGFR status unknown; duration of response, 7.5 months); two patients had stable disease.
Conclusions
With C4 cut-off and prophylactic bile acid sequestrant implementation, INCB062079 demonstrated a manageable safety profile and evidence of target inhibition. In view of the rarity of FGF19/FGFR4 alterations and slow patient accrual, the study was terminated before establishing an MTD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In this phase I study of INCB062079 for previously treated advanced solid tumors, INCB062079 demonstrated a manageable safety profile, generally dose-proportional pharmacokinetics, and pharmacodynamics consistent with target inhibition. |
Because of slow patient accrual, the study was terminated before a maximum tolerated dose of INCB062079 could be identified, and clinical development was terminated. |
Nevertheless, the results validate INCB062079 as a clinical grade fibroblast growth factor receptor-4 inhibitor and support future research into the relevance of the FGF19/FGFR4 signaling axis as a therapeutic target in solid tumors. |
1 Introduction
Fibroblast growth factor (FGF)-19 is an endocrine factor produced by the ileum in response to postprandial luminal bile salts [1,2,3]. In the liver, FGF19 binds to FGF receptor (FGFR)-4, and the co-receptor βklotho, thereby regulating both bile salt formation and hepatocyte proliferation [3,4,5]. Under physiologic conditions, FGF19 and its receptors are thought to play a critical role in protecting against cholestatic liver injury [1].
Emerging evidence indicates that dysregulation of the FGF19/FGFR4 signaling axis contributes to oncogenesis in hepatocellular carcinoma (HCC) and several other solid tumors [6,7,8]. In clinical HCC samples, FGF19 is genomically amplified in the context of an 11q13 amplicon in approximately 5% of tumors [9, 10], is upregulated by epigenetic mechanisms in approximately 23% of tumors [7], and is overexpressed in 27% of patients by immunohistochemistry [7]. Possible activating alterations in FGFR4 as well as FGF19 amplifications have been reported in several other cancer types, including gastric, liver, lung, uterine, bladder, ovarian, nasopharyngeal, colorectal, breast cancers, rhabdomyosarcoma, and head and neck squamous cell carcinoma among others [8, 11,12,13]. In a small tumor series (N = 199), patients with intrahepatic or perihilar cholangiocarcinoma with above average FGFR4 expression had significantly poorer prognosis than patients with below average FGFR4 expression [14]. FGFR4 has also been reported to be overexpressed in rhabdomyosarcoma [15] and in high-grade serous ovarian tumors, where expression was demonstrated to be significantly higher when compared with normal ovarian and fallopian tube tissue [13].
Available preclinical evidence also indicates that FGF19 autocrine or paracrine secretion drives uncontrolled cellular proliferation, growth, and malignant transformation in HCC. Experiments performed in vitro and in vivo indicate that blocking FGF19 with monoclonal antibodies or inhibition of FGFR4 with small molecule inhibitors results in growth inhibition or tumor shrinkage of FGF19-amplified HCCs [16,17,18,19,20,21,22]. In the clinic, the orally bioavailable, selective FGFR4 inhibitor fisogatinib (BLU-554) was associated with an overall response rate (ORR) of 17% in 66 patients with FGF19-positive HCC (identified using immunohistochemistry), illustrating the potential therapeutic utility of targeting FGFR4 in HCC [7].
INCB062079 is a potent, selective, orally bioavailable, irreversible inhibitor of FGFR4 [21]. In vitro, INCB062079 inhibits FGFR4 at low nanomolar concentrations, blocks downstream signaling, and inhibits proliferation in FGF19-amplified and FGFR4-mutant cell lines. INCB062079 also inhibits growth in several tumor models driven by aberrant FGF19 or FGFR4 signaling at tolerated doses in preclinical studies [21].
Based on these preclinical and early-stage clinical data and because of the unmet treatment needs of patients with advanced solid tumors, a first-in-human study of INCB062079 was conducted in this patient population and is described herein.
2 Methods
2.1 Study Design
This was an open-label, dose-escalation (Part 1) and dose-expansion (Part 2) study of INCB062079 in patients with previously treated advanced solid tumors. The primary objective was to determine the safety, tolerability, and dose-limiting toxicity (DLT) and maximum tolerated dose (MTD) of INCB062079. Secondary objectives were to assess the preliminary efficacy of INCB062079 in terms of ORR in patients with measurable disease, and to evaluate the pharmacokinetics (PK) and pharmacodynamics (PD) of INCB062079, as assessed by plasma FGF19 and serum bile acid concentrations, including concentrations of the bile acid synthesis precursor 7α-hydroxy-4-cholesten-3-one (C4).
In Part 1, eligible patients received INCB062079 orally once daily or twice daily continuously over a 28-day cycle. The starting dose for Cohort 1 was set at 10 mg once daily based on one-sixth of the highest nonseverely toxic dose observed in preclinical toxicology per the US Food and Drug Administration guidelines [23]. Intrapatient dose escalation was not allowed. Dose escalation followed standard 3 + 3 rules, and the decision to open a dose-escalation cohort was made by a safety review committee comprising the study sponsor and academic investigators. All patients continued treatment until progression of disease, intolerable toxicity, or consent withdrawal.
In Part 2, separate dose-expansion cohorts at the MTD in Part 1 were planned for HCC patients with and without FGF19 amplification, as well as a basket cohort for patients with bile duct cancer or any solid tumor with known activating alterations in FGFR4 or FGF19.
The study, registered under ClinicalTrials.gov (NCT03144661), was performed in accordance with the International Conference for Harmonisation Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable local regulations with approval from institutional review boards. The study protocol was reviewed and approved by the respective institutional review boards of the participating institutions (Online Supplementary Table 1). Written informed consent was obtained for all patients before performing study-related procedures.
2.2 Patients
Eligible patients were men and women ≥ 18 years of age who had any solid tumor malignancy, as long as they had documented FGF19/FGFR4 alterations. Patients with histology confirmed HCC, cholangiocarcinoma, esophageal, nasopharyngeal, or serous ovarian cancer were eligible regardless of FGF19/FGFR4 alterations. FGF19/FGFR4 alterations were assessed based on next-generation sequencing, either centrally (FoundationOne®, Foundation Medicine Inc. Cambridge, MA, USA) or locally. FGF19/FGFR4 alterations included, but were not limited to, FGFR4 amplification, FGFR4-activating mutations, and FGF19 amplification. Patients must also have progressed after prior therapy and either have no further effective standard anticancer therapy available to them (including patient refusal) or be intolerant to standard anticancer therapy. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1, a life expectancy of > 12 weeks, intact organ function, and no history of hepatic encephalopathy. Patients with Child–Pugh B or C liver function were excluded. Archival tumor tissue or a fresh biopsy was required for study enrollment.
Key exclusion criteria included prior receipt of selective FGFR4 inhibitors within the last 6 months, receipt of systemic anticancer therapy within 28 days of study dosing, active or untreated central nervous system metastasis, or other systemic illness that could interfere with study participation.
During the conduct of the study, the protocol was amended to require all newly enrolled patients to have a C4 concentration < 40.9 ng/mL and to take a prophylactic bile acid sequestrant for the prevention of diarrhea [24] while receiving INCB062079 treatment. The threshold C4 value of 40.9 ng/mL corresponded to the mean plus two standard deviations of serum C4 concentration data derived from an analysis of patients who had been enrolled before the protocol amendment date. Serum C4 concentrations were determined using a validated liquid chromatography–tandem mass spectrometry assay.
2.3 Clinical Assessments
Patients were screened for medical history and demographics. Comprehensive laboratory tests were performed on days 1, 2, and 15 of cycle 1 and days 1 and 15 of each subsequent cycle. On days 1, 2, 8, 15, and 16 of cycle 1 and day 1 of each subsequent 28-day cycle, patients were assessed on prior/concomitant medication review, physical examination, vital signs, and adverse events. Adverse events were recorded, graded, and attributed using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. Cross-sectional imaging was conducted with computed tomography or magnetic resonance imaging of the chest, abdomen (liver 3 or 4 phase for HCC), and pelvis every 8 weeks. Antitumor activity was assessed by investigators using modified Response Evaluation Criteria in Solid Tumors (RECIST) [25] for HCC and RECIST v 1.1 [26] for other tumors.
2.4 Pharmacokinetic and Pharmacodynamic Assessments
Trough (predose) fasting PK samples were drawn approximately 1 h before INCB062079 administration during cycle 1 on days 1, 2, 8, 15, and 16. Patients continued to fast until 1 h after taking INCB062079. During cycle 1, on day 1 and day 15, additional PK samples were drawn at 0.5, 1, 2, 4, 6, and 8 h postdose. Plasma samples were analyzed for INCB062079 using a validated liquid chromatography‒tandem mass spectrometry assay. Urine samples were collected predose on cycle 1 day 15 and then following INCB062079 administration, over an 8-h interval (total urine). See Online Supplementary Methods for additional PK methodologies.
Plasma concentrations of FGF19 and serum bile acids (analytes directly related to FGFR4 pathway modulation) were assayed for changes directly related to drug exposure. PD samples were drawn at the same timepoints as the PK samples (above). Correlative markers, including FGF19, C4, and bile acids, were analyzed in plasma samples collected predose during cycle 1 day 1 and cycle 1 day 15 and any time during day 1 of cycles 2, 4, 8, and 12 to assess associations between response to INCB062079 treatment and changes from baseline in plasma concentrations of FGF19, C4, and bile acids.
2.5 Dose-Limiting Toxicity Criteria
DLT was defined as the occurrence of any grade 3 or higher nonhematologic toxicity, except transient (duration ≤ 72 h) abnormal laboratory values without associated clinically significant signs or symptoms; grade 3 nausea, vomiting, and diarrhea adequately controlled with medical therapy within 48 h; or changes in cholesterol and triglycerides. Liver function test DLTs were defined as aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) concentrations > 5.0 and < 20 × upper limit of normal (ULN) lasting > 7 days or recurring upon rechallenge, or AST and/or ALT concentrations ≥ 20 × ULN or ALT elevations consistent with Hy's law (ALT concentration > 3 × ULN with concurrent serum total bilirubin elevation to > 2 × ULN without cholestasis [serum alkaline phosphatase [27] < 2 × ULN] or other explanations for increased ALT and total bilirubin levels). Grade 4 or higher hematologic toxicity, grade 3 thrombocytopenia associated with bleeding, or febrile neutropenia were also considered DLTs. DLTs were assessed only during the first 28 days of treatment. See Online Supplementary Table 2 for further details of DLT definitions. Patients who had dose reductions (but not meeting DLT criteria) during the first cycle who had not received at least 75% of the prescribed dose for that cohort were not considered evaluable for the purposes of determining the MTD. See Online Supplementary Table 2 for further details.
2.6 Statistical Analysis
All data were reported and tabulated using descriptive statistics. The efficacy and safety populations included all patients who received at least one dose of study drug. The PK-evaluable population included patients who received at least one dose of study drug and provided at least one sample for plasma PK. The MTD was defined as the one dose level below that at which one-third or more of patients in a particular cohort report a DLT. PK parameters were calculated from blood plasma concentrations using standard noncompartmental (model-independent) methods. Linear regression was used to evaluate the relationship between INCB062079 exposure and FGF19 and bile salt concentrations.
3 Results
3.1 Patients
The study was conducted from the date the first patient was enrolled and received their first dose (22 June 2017) to the date that the last patient completed the study (24 June 2020). Fifty-three patients were screened for eligibility (Fig. 1); six patients withdrew consent and 22 were excluded for not meeting the eligibility criteria, including four who had C4 concentrations ≥ 40.9 ng/mL and were excluded after the protocol amendment requiring patients to have C4 concentrations < 40.9 ng/mL. The remaining 25 patients were enrolled across three study sites in Belgium and five sites in the United States. Among these 25 patients, two did not receive study treatment and were not assigned to dose cohorts; 23 patients were therefore included for assessment of safety and efficacy.

Consort diagram. aFour patients with plasma C4 concentrations ≥ 40.9 ng/mL were excluded based on a protocol amendment to require all newly enrolled patients to have a C4 concentration < 40.9 ng/mL and to take a prophylactic bile acid sequestrant for the prevention of diarrhea while receiving INCB062079 treatment. BID twice daily, C4 7α-hydroxy-4-cholesten-3-one, QD once daily
Patients had a median age of 57 years (range 29–75) and 56.5% were female; patients were predominantly White (78.3%) and had Eastern Cooperative Oncology Group performance status of 1 (87.0%) [Table 1]. The most common advanced/metastatic cancer types included hepatobiliary cancers (47.8%; HCC n = 4, cholangiocarcinoma n = 7) and ovarian cancer (39.1%, n = 9). Patients had a median of two (range 0–9) prior systemic treatments and two patients had received prior treatment with an FGFR1-3 inhibitor. FGF/FGFR alterations were identified in two patients (8.7%), absent in nine patients (39.1%), and unknown in 12 patients (52.2%). Of the two patients with known FGF/FGFR alterations, one had cholangiocarcinoma with FGF3, FGF4, and FGF19 amplifications, and the other had esophageal cancer with FGF3, FGF4, FGF10, and FGF19 amplifications and an FGFR4 p.S551F mutation.
3.2 Safety and Tolerability
In total, five dosing cohorts were examined sequentially: Cohort 1: 10 mg once daily; Cohort 2: 10 mg twice daily; Cohort 3: 15 mg twice daily; and Cohorts 4 and 5 (10 mg and 15 mg twice daily, respectively, which required concurrent treatment with bile acid sequestrants and baseline C4 < 40.9 ng/mL). Two of the six patients enrolled in Cohort 3 experienced one DLT each. One patient had grade 3 diarrhea lasting over 48 h despite supportive treatment; this event resolved after study drug interruption and treatment with bile acid sequestrant (cholestyramine) and antidiarrheal therapy (loperamide and diphenoxylate/atropine). The second patient developed grade 3 increased ALT and AST—these events recurred after rechallenge of INCB062079 at a reduced dose of 10 mg twice daily. Retrospective analysis indicated that baseline C4 concentrations in these two patients were the highest among all patients studied. Study entry criteria were therefore modified to require prophylactic bile acid sequestrants for the prevention of diarrhea and to exclude patients with baseline C4 concentrations ≥ 40.9 ng/mL. Implementation of this protocol amendment allowed dosing at both the 10 and 15 mg twice-daily doses without additional DLTs; however, higher doses were not assessed and dose expansion (Part 2) was not conducted because the study was terminated.
Among the 23 patients in the safety population, 22 (95.7%) reported at least one treatment-emergent adverse event (TEAE) and 21 (91.3%) reported a TEAE that was considered related to INCB062079. The most common any-grade TEAEs occurring in over 30% of patients were diarrhea (60.9%), fatigue (56.5%), nausea (47.8%), abdominal pain (39.1%), and decreased appetite (34.8%) (Table 2). The most common treatment-related adverse events occurring in over 30% of patients were diarrhea (60.9%), fatigue (39.1%), and nausea (30.4%).
Thirteen patients (56.5%) had at least one grade 3 or higher TEAE, the most common being dyspnea and hypokalemia (13.0% each). Four patients had TEAEs with a fatal outcome: one patient (10 mg twice daily) had grade 4 pulmonary embolism; one patient (10 mg twice daily) had grade 4 bowel obstruction; one patient (10 mg twice daily) had grade 4 dyspnea; and one patient (15 mg twice daily) had grade 4 acute hepatic failure and acute kidney injury. The latter two patients were enrolled after the protocol amendment required prophylactic bile acid sequestrants and C4 concentrations < 40.9 ng/mL. None of the fatal TEAEs were considered related to INCB062079 treatment by the investigators.
Overall, patients were treated for a median two cycles (range 1–35); median treatment duration was 55 days (range 6–955). Median average daily dose of INCB062079 was 19.5 mg (range 9.8–30.4 mg/day). Dose was reduced in two patients (8.7%) owing to TEAEs (grade 3 diarrhea, n = 1; grade 3 AST and ALT increased, n = 1). Seven patients (30.4%) had dose interruption in response to a TEAE. TEAEs leading to dose interruption in more than one patient were diarrhea (two patients, grade 1 and grade 2, respectively) and fatigue (two patients, both grade 3). One patient discontinued due to a gastrointestinal obstruction TEAE that was unrelated to treatment.
3.3 Pharmacokinetics
INCB062079 plasma concentration versus time curves following treatment on cycle 1 day 1 and cycle 1 day 15 (steady-state) are shown in Fig. 2. With multiple-dose administration in fasting patients, the maximum plasma concentration of INCB062079 typically occurred 2–3 h postdose and subsequently exhibited a monophasic decay, with a steady-state geometric mean half-life of approximately 6 h that was not dose-dependent (Online Supplementary Table 3). Exposure was generally dose proportional. INCB062079 was associated with a low clearance at steady state (CLss/F; geometric mean 2.95–4.99 L/h) and a low volume of distribution (Vz/F; geometric mean 30.7–36.5 L). Steady-state exposure to INCB062079 was higher when bile acid sequestrants were used, although this observation may be due to a limited number of patients evaluated for PK (Online Supplementary Table 3). In an exploratory analysis of urine from 12 patients, the fraction of INCB062079 excreted in urine was 0.663% (range 0.305–2.63%), with a geometric mean renal clearance of 0.024 L/h, indicating renal clearance plays a minor role in total systemic clearance.

Plasma INCB062079 concentrations (mean ± SE) in patients following oral administration of INCB062079 during a C1D1 and b C1D15 (steady state). *Because the BID dosing schedule consisted of two doses of INCB062079 administered within a 24-h period, plasma concentration data were collected up to 24 h following QD dosing only. BID twice daily, C cycle, D day, IC50 INCB062079 concentration resulting in 50% inhibition of FGFR4 activity, IC90 INCB062079 concentration resulting in 90% inhibition of FGFR4 activity, QD once daily, SE standard error
3.4 Pharmacodynamics
The mean change from baseline in serum bile acid concentration increased with increasing area under the INCB062079 plasma concentration versus time curve (AUC) at steady state, and the relationship was statistically significant (p value of slope = 0.02) [Fig. 3a]. A positive correlation was observed for INCB062079 steady-state AUC and C4 concentration change from baseline AUC at cycle 1 day 15, and the relationship was statistically significant (p value of slope < 0.001) [Fig. 3b]. Changes in plasma FGF19 concentrations demonstrated a response to FGFR4 inhibition following INCB062079 treatment (Fig. 3c, d). Plasma FGF19 concentrations were elevated at all dosages, with the highest fold increase at 10 mg twice daily at cycle 1 day 15 (4.5-fold) [Fig. 3c].

Steady-state AUC of INCB062079 versus mean change from baseline after C1D15 following INCB062079 treatment in a serum bile acid concentration; b plasma C4 AUC for INCB062079 at C1D15 (steady-state); c fold-change in FGF19 plasma concentrations from C1D1 to C1D15; d fold-change in FGF19 plasma concentrations from C1D1 to C2D1. Symbols denote values for the individual patients who had FGF19 plasma concentration data available at the relevant timepoints. Red symbols denote patients with hepatocellular carcinoma. aIn panel d, FGF19 plasma concentration data were unavailable for one patient receiving 10 mg QD and one patient receiving 15 mg BID. AUC area under the plasma concentration-time curve, AUCss,0–24h area under the plasma concentration-time curve from 0 to 24 h at steady-state, BID twice daily, C cycle, C4 7α-hydroxy-4-cholesten-3-one, D day, QD once daily
3.5 Antitumor Activity
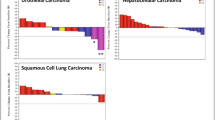
Of the 23 patients analyzed, none attained a complete response, one patient receiving 15 mg twice daily achieved a best overall response of partial response, two achieving stable disease and 16 having progressive disease. One patient was not evaluable for response and three patients had missing postbaseline assessments. The ORR by RECIST v1.1 was 4.3% (95% confidence interval [CI] 0.11–21.95). The patient achieving a partial response had advanced endometrioid ovarian carcinoma with unknown FGF19/FGFR4 status, had previously undergone exploratory laparotomy with gastrocolic/infrasonic omentectomy and bilateral salpingo-oophorectomy, and had received four prior lines of cytotoxic chemotherapy. This patient received INCB062079 15 mg twice daily and had a best percentage change from baseline of 65.1% reduction in target lesion size, with a duration of response of 7.5 months (Fig. 4a, b). One of the two patients achieving stable disease had advanced HCC and unknown FGF19 status, and had progressed on sorafenib before study enrollment. This patient, who received INCB062079 10 mg once daily, achieved stable disease without progression for 31.3 months. The other patient achieving stable disease had intrahepatic cholangiocarcinoma with no reported FGF19 status and had been previously treated with surgery, radiation, and five lines of systemic therapy. This patient received INCB062079 10 mg twice daily and achieved stable disease without progression for 4.9 months.

a Waterfall plot of best percentage change in the sum of target lesions for individual patients receiving INCB062079 (efficacy-evaluable population). Data were from investigator review per standard RECIST v1.1 and modified RECIST for hepatocellular carcinoma, and response was unconfirmed. b Duration of treatment and response assessments among individual patients receiving INCB062079. aThis patient had cholangiocarcinoma and FGF3, FGF4, and FGF19 amplifications. bThis patient had esophageal cancer and FGF3, FGF4, FGF10, and FGF19 amplifications, and an FGFR4 p.S551F mutation. cThis patient had ovarian cancer. dPatients were enrolled after a protocol amendment requiring all newly enrolled patients to have a C4 concentration < 40.9 ng/mL and to take a prophylactic bile acid sequestrant for the prevention of diarrhea while receiving INCB062079 treatment. BID twice daily, C4 7α-hydroxy-4-cholesten-3-one, GE gastroesophageal, NE not evaluable, PD progressive disease, PR partial response, QD once daily, RECIST Response Evaluation Criteria in Solid Tumors, SD stable disease
4 Discussion
Dysregulation of FGF19/FGFR4/βklotho-mediated signal transduction through the MAPK, PI3K/AKT/mTOR, JAK-STAT, NFκB, and Wnt/β-catenin pathways can result in uncontrolled cellular growth in preclinical models of HCC [6, 8, 28]. Whereas this oncogenic mechanism is highly relevant to hepatobiliary cancers, dysregulation of the FGF19/FGFR4/βklotho signaling axis has also been observed in other solid tumors [6,7,8]. We therefore evaluated INCB062079, a potent, oral, irreversible inhibitor of FGFR4, in patients with solid tumors, including hepatobiliary cancers, in the context of a first-in-human clinical trial. Manageable on-target gastrointestinal toxicity was observed in most patients at the initial dosages. However, dose escalation beyond 15 mg twice daily was not possible because two of six patients experienced gastrointestinal/hepatobiliary DLTs. The protocol was subsequently amended to include eligibility criteria that mitigated this toxicity by requiring the prophylactic use of bile acid sequestrants and a pretreatment serum C4 concentration cut-off < 40.9 ng/mL. This amendment resulted in an apparent blunting of toxicity allowing successful dose escalation. Importantly, the generally dose-proportional PK of INCB062079 was associated with an expected increase in plasma C4 and FGF19 concentrations, consistent with target inhibition of FGFR4 in vivo. A partial response was observed at the highest dose levels tested (15 mg twice daily), indicating a signal of antitumor activity. Despite these findings, because of the rarity of FGF19/FGFR4 alterations and the slow accrual of patients who satisfied the eligibility criteria, the study was terminated before the MTD could be identified, and the clinical development of INCB062079 was terminated.
Increasing postprandial luminal bile salt concentrations result in exocrine farnesoid X receptor (FXR)-induced production of FGF19 by enterocytes, which serves to activate FGFR4/βklotho-mediated signaling in hepatocytes and cholangiocytes [27, 29]. Under normal physiologic conditions, FGF19/FGFR4/βklotho signal activation suppresses bile acid biosynthesis in hepatocytes by downregulating the CYP7A1 gene [3, 4, 8] via SHP/liver receptor homolog-1 (LRH-1)-dependent [30, 31] and JNK-dependent [4] feedback mechanisms. Consistent with this, FGFR4 inhibition was associated with increased CYP7A1 RNA expression and bile salt excretion in cynomolgus monkeys [21]. Furthermore, in murine models, knocking out FGFR4 or βklotho leads to elevated bile acid excretion and an increased bile acid pool [5, 32]. Moreover, βklotho–/– mice have been shown to display phenotypic characteristics suggestive of early-stage liver damage, including increased liver size, increased ALT and AST levels, increased proinflammatory cytokine expression, and fibrogenesis [33]. It was further demonstrated that these characteristics were associated with a large increase in microbiota-derived deoxycholic acid [33]. Given the potential toxicity of excessive bile salt accumulation [33], the expected on-target gastrointestinal and hepatobiliary toxicity of selective FGFR4 inhibitors must be scrutinized and mitigated to ensure an adequate therapeutic index of this drug class. The observed safety profile of INCB062079 was consistent with its proposed mechanism of action, with manageable any-grade diarrhea (60.9%), nausea (47.8%), vomiting (26.1%), and AST and ALT increases (17.4 and 13.0%, respectively). Such on-target effects were also observed in 106 patients with advanced HCC receiving the selective FGFR4 inhibitor fisogatinib in a recent phase I study [7], which reported treatment-related adverse events of diarrhea (any-grade 74%; grade 3 or higher, 8%), nausea (42%; 2%), and vomiting (35%; 4%). Treatment-related grade 3 or higher ALT and AST increase adverse events were also observed in 11% and 15%, respectively, of patients treated with fisogatinib [7]. Because this class of drug has greatest applicability in hepatobiliary cancers, the potential liability of on-target FGFR4 toxicity and need for mitigation efforts are of clear importance.
Without mitigation efforts, the INCB062079 dose could not be escalated without intolerable dose-limiting gastrointestinal toxicity in patients, indicative of a narrow therapeutic index. For this reason, efforts were made to select patients with low baseline plasma C4 expression and prophylactically treat with bile acid sequestrants. Preclinical toxicology data indicate that bile acid sequestration is a novel method to prevent FGFR4-induced hepatic and gastrointestinal toxicity [34, 35]. Although the patient sample sizes in the present study are too small to draw conclusions, diarrhea was reported by a smaller percentage of patients who received concurrent prophylactic treatment with a bile acid sequestrant (Cohorts 4 and 5), compared with patients who did not receive such treatment. Bile acid sequestrant treatment may therefore have merit as a clinical strategy to blunt gastrointestinal toxicity associated with FGFR4 inhibition and should be explored in future studies.
Importantly, INCB062079 exhibited linear exposure relative to dose and did not accumulate with repeat dosing. The addition of bile acid sequestrant did not appear to interfere with absorption or reduce INCB062079 exposure. PK/PD modeling did indicate a statistically significant increase in bile salt and C4 concentrations with increasing INCB062079 exposure and a two- to four-fold increase in FGF19 concentrations with repeat dosing. These findings suggest that INCB062079 inhibits its proposed target, further supporting INCB062079 as a clinical grade FGFR4 inhibitor. FGF19/FGFR4/βklotho pathway inhibition by INCB062079 in tumor tissues could not be confirmed because paired pretreatment and on-treatment biopsies were not obtained in this study (per protocol).
Determination of antitumor activity was not a primary objective of this phase I study and could not be satisfactorily assessed given that higher INCB062079 doses were not tested. Interestingly, a partial response was seen in a patient with ovarian cancer with unknown FGF19/FGFR status. In addition, long-term stable disease was observed in a patient with HCC treated with INCB062079 (31.3 months). This observation is in keeping with previous findings with fisogatinib, where eight patients with HCC achieved either partial or complete responses lasting over 6 months [7]. Taken together, these observations are hypothesis-generating and support further investigation of the efficacy and safety of selective FGFR4 inhibitors in patients with advanced HCC and other solid tumors. No correlation was established between potential biomarkers and clinical efficacy; the two patients with known FGF19 amplifications did not respond to study treatment. Other selective FGFR4 inhibitors currently in phase I/II clinical trials for HCC or other advanced solid tumors as monotherapy or in combination with other anticancer agents include roblitinib (FGF401) [22] (NCT02325739), EVER4010001 (NCT04699643), ABSK-011 [36] (NCT04906434), and H3B-6527 [37] (NCT02834780). Together, these studies will help clarify the merit of selective FGFR4 inhibitor treatments in patients with solid tumors.
Limitations of the study include a lack of pretreatment data on the types of FGF19/FGFR4 alterations, including fusions or other rearrangements, mutations, amplifications, as well as FGF19 protein overexpression. In addition, FGFR4/FGF19 genomic testing data were missing in 52.2% of patients in the dose-escalation cohort; however, this reflects the design of Part 1, which enrolled patients regardless of FGFR4/FGF19 alteration status. Expansion cohorts had been planned for these analyses to explore the predictive and prognostic significance of FGF19 and FGFR4 alterations on patient outcome. Importantly, inclusion of other histology/tumor types led to hypothesis-generating clinical observations that would have been explored in the planned expansion cohorts. For example, the only partial responder in the present study had heavily pretreated ovarian cancer. Recent retrospective and translational analyses indicate that FGFR4 overexpression is associated with a worse outcome in advanced ovarian cancer, and that blockade of ligand-dependent FGFR4 activation is deleterious to ovarian cancer cell growth in vitro and in vivo [13]. Finally, although only a minority of patients had C4 concentrations exceeding the C4 concentration cut-off initiated with the protocol amendment, this additional eligibility criterion served to slow accrual and thus further narrowed a pool of patients with an already infrequent FGF19/FGFR4 variant. Indeed, the C4 threshold was established based on a limited sample size using an assay not certified by the Clinical Laboratory Improvement Amendments (CLIA) while the study was ongoing, without statistical power for robust analysis. In retrospect, C4 levels should be embedded as an integrated biomarker of toxicity. Nevertheless, the data presented herein help to credential INCB062079 as a bona fide clinical grade inhibitor of FGFR4, provide useful information for future drug development of agents blocking FGFR4, and support continued investigation to determine the relevance of the FGF19/FGFR4 axis as a target in solid tumors.
References
Luo J, Ko B, Elliott M, Zhou M, Lindhout DA, Phung V, et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med. 2014;6:247ra100. https://doi.org/10.1126/scitranslmed.3009098.
Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25. https://doi.org/10.1016/j.cmet.2005.09.001.
Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530–6. https://doi.org/10.1111/j.1365-2796.2006.01731.x.
Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–91. https://doi.org/10.1101/gad.1083503.
Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y, Nabeshima Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking βKlotho. J Clin Invest. 2005;115:2202–8. https://doi.org/10.1172/JCI23076.
Prieto-Dominguez N, Shull AY, Teng Y. Making way for suppressing the FGF19/FGFR4 axis in cancer. Future Med Chem. 2018;10:2457–70. https://doi.org/10.4155/fmc-2018-0099.
Kim RD, Sarker D, Meyer T, Yau T, Macarulla T, Park JW, et al. First-in-human phase I study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 2019;9:1696–707. https://doi.org/10.1158/2159-8290.CD-19-0555.
Levine KM, Ding K, Chen L, Oesterreich S. FGFR4: a promising therapeutic target for breast cancer and other solid tumors. Pharmacol Ther. 2020;214: 107590. https://doi.org/10.1016/j.pharmthera.2020.107590.
Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58:706–17. https://doi.org/10.1002/hep.26402.
Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–11. https://doi.org/10.1038/ng.3252.
Shi S, Li X, You B, Shan Y, Cao X, You Y. High expression of FGFR4 enhances tumor growth and metastasis in nasopharyngeal carcinoma. J Cancer. 2015;6:1245–54. https://doi.org/10.7150/jca.12825.
Gao L, Lang L, Zhao X, Shay C, Shull AY, Teng Y. FGF19 amplification reveals an oncogenic dependency upon autocrine FGF19/FGFR4 signaling in head and neck squamous cell carcinoma. Oncogene. 2019;38:2394–404. https://doi.org/10.1038/s41388-018-0591-7.
Zaid TM, Yeung TL, Thompson MS, Leung CS, Harding T, Co NN, et al. Identification of FGFR4 as a potential therapeutic target for advanced-stage, high-grade serous ovarian cancer. Clin Cancer Res. 2013;19:809–20. https://doi.org/10.1158/1078-0432.CCR-12-2736.
Xu YF, Yang XQ, Lu XF, Guo S, Liu Y, Iqbal M, et al. Fibroblast growth factor receptor 4 promotes progression and correlates to poor prognosis in cholangiocarcinoma. Biochem Biophys Res Commun. 2014;446:54–60. https://doi.org/10.1016/j.bbrc.2014.02.050.
Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–9. https://doi.org/10.1038/89044.
Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015;5:424–37. https://doi.org/10.1158/2159-8290.CD-14-1029.
Joshi JJ, Coffey H, Corcoran E, Tsai J, Huang CL, Ichikawa K, et al. H3B–6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res. 2017;77:6999–7013. https://doi.org/10.1158/0008-5472.CAN-17-1865.
Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell. 2011;19:347–58. https://doi.org/10.1016/j.ccr.2011.01.040.
French DM, Lin BC, Wang M, Adams C, Shek T, Hotzel K, et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE. 2012;7: e36713. https://doi.org/10.1371/journal.pone.0036713.
Liu PCC, Lu L, Bowman K, Stubbs MC, Wu L, DiMatteo D, et al. Selective inhibition of FGFR4 by INCB062079 is efficacious in models of FGF19- and FGFR4-dependent cancers [abstract]. Cancer Res. 2017;77:2100. https://doi.org/10.1158/1538-7445.Am2017-2100.
Ruggeri B, Stubbs M, Yang Y-O, Juvekar A, Lu L, Condon S, et al. The novel FGFR4-selective inhibitor INCB062079 is efficacious in models of hepatocellular carcinoma harboring FGF19 amplification [abstract]. Cancer Res. 2017;77:1234. https://doi.org/10.1158/1538-7445.Am2017-1234.
Weiss A, Adler F, Buhles A, Stamm C, Fairhurst RA, Kiffe M, et al. FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol Cancer Ther. 2019;18:2194–206. https://doi.org/10.1158/1535-7163.MCT-18-1291.
US Food and Drug Administration. Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers. Accessed 29 Oct 2021.
Scaldaferri F, Pizzoferrato M, Ponziani FR, Gasbarrini G, Gasbarrini A. Use and indications of cholestyramine and bile acid sequestrants. Intern Emerg Med. 2013;8:205–10. https://doi.org/10.1007/s11739-011-0653-0.
Lencioni R, Llovet JM. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin Liver Dis. 2010;30:52–60. https://doi.org/10.1055/s-0030-1247132.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. https://doi.org/10.1016/j.ejca.2008.10.026.
Jung D, York JP, Wang L, Yang C, Zhang A, Francis HL, et al. FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch. 2014;466:1011–9. https://doi.org/10.1007/s00424-013-1364-3.
Chen Z, Jiang L, Liang L, Koral K, Zhang Q, Zhao L, et al. The role of fibroblast growth factor 19 in hepatocellular carcinoma. Am J Pathol. 2021;191:1180–92. https://doi.org/10.1016/j.ajpath.2021.04.014.
Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR-FGF15/19 pathway. Dig Dis. 2015;33:327–31. https://doi.org/10.1159/000371670.
Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–26. https://doi.org/10.1016/s1097-2765(00)00051-4.
Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–15. https://doi.org/10.1016/s1097-2765(00)00050-2.
Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–9. https://doi.org/10.1074/jbc.275.20.15482.
Somm E, Henry H, Bruce SJ, Bonnet N, Montandon SA, Niederländer NJ, et al. β-Klotho deficiency shifts the gut-liver bile acid axis and induces hepatic alterations in mice. Am J Physiol Endocrinol Metab. 2018;315:E833–47. https://doi.org/10.1152/ajpendo.00182.2018.
Bartz R, Fukuchi K, Ohtsuka T, Lange T, Gruner K, Watanabe I, et al. Preclinical development of U3–1784, a novel FGFR4 antibody against cancer, and avoidance of its on-target toxicity. Mol Cancer Ther. 2019;18:1832–43. https://doi.org/10.1158/1535-7163.MCT-18-0048.
Schadt HS, Wolf A, Mahl JA, Wuersch K, Couttet P, Schwald M, et al. Bile acid sequestration by cholestyramine mitigates FGFR4 inhibition-induced ALT elevation. Toxicol Sci. 2018;163:265–78. https://doi.org/10.1093/toxsci/kfy031.
Chen Z. Discovery and characterization of a novel FGFR4 Inhibitor for the treatment of hepatocellular carcinoma [abstract]. Cancer Res. 2018;78:LB-272. https://doi.org/10.1158/1538-7445.Am2018-lb-272.
Macarulla T, Moreno V, Chen LT, Sawyer MB, Goyal L, Muñoz Martín AJ, et al. Phase I study of H3B–6527 in hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (ICC). J Clin Oncol. 2021;39:4090. https://doi.org/10.1200/JCO.2021.39.15_suppl.4090.
Acknowledgements
The authors thank the patients and families, investigators, and site personnel who participated in this study. This study was sponsored by Incyte Corporation (Wilmington, DE, USA). Medical writing assistance was provided by Simon J. Slater, PhD, CMPP, of Envision Pharma Group (Philadelphia, PA, USA), and funded by Incyte Corporation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was funded by Incyte Corporation and, in part, through the NIH/NCI Cancer Center Support Grant P30 CA008748 (JJH).
Conflict of interest
James J. Harding: Consulting or advisory role for Adaptimmune, Bristol Myers Squibb, CytomX, Eisai, Eli Lilly & Company, Exelixis, Imvax, Merck, Zymeworks; and research funding from Boehringer Ingelheim (Inst), Bristol Myers Squibb (Inst), Calithera Biosciences (Inst), Debiopharm (Inst), Eli Lilly & Company (Inst), Incyte Corporation (Inst), Novartis (Inst), Pfizer (Inst), Polaris Group (Inst), Yiviva (Inst), and Zymeworks (Inst). Christiane Jungels: Travel, accommodations, and expenses from Ipsen, PharmaMar. Jean-Pascal Machiels: Consulting or advisory role for ALX Oncology, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, CUE Biopharma, Debiopharm, Incyte Corporation, Innate Pharma, Janssen, Merck Serono, Merck Sharp & Dohme, Nanobiotix, Novartis, Pfizer, Roche; research funding from Bayer, Janssen, Novartis, Sanofi; and travel, accommodations, and expenses from Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, and Pfizer. David C. Smith: Research funding from Agensys (Inst), Atterocor (Inst), Bayer (Inst), Boston Biomedical (Inst), Bristol Myers Squibb/Medarex (Inst), Celgene (Inst), Eli Lilly & Company (Inst), ESSA (Inst), Exelixis (Inst), Genentech (Inst), ImClone Systems (Inst), Incyte Corporation (Inst), MedImmune (Inst), Medivation/Astellas Pharma (Inst), Millennium Pharmaceuticals (Inst), Novartis (Inst), OncoGenex (Inst), OncoMed Pharmaceuticals (Inst), Regeneron (Inst), SeaGen (Inst), Takeda (Inst), Tekmira Pharmaceuticals (Inst), and Teva Pharmaceuticals (Inst). Chris Walker, Tao Ji, Pin Jiang: Former employees of Incyte Corporation. Ekaterine Asatiani, Xin Li: Employment by, and stock ownership in, Incyte Corporation. Eric Van Cutsem: Consulting or advisory role for Array BioPharma, AstraZeneca, Bayer, Biocartis, Bristol Myers Squibb, Celgene, Daiichi Sankyo, Eli Lilly & Company, GlaxoSmithKline, Halozyme, Incyte Corporation, Merck KGaA, Merck Sharp & Dohme, Novartis, Pierre Fabre, Roche, Servier, Sirtex Medical, and Taiho Pharmaceutical Co., Ltd; and research funding from Amgen (Inst), Bayer (Inst), Boehringer Ingelheim (Inst), Bristol Myers Squibb (Inst), Celgene (Inst), Eli Lilly & Company (Inst), Ipsen (Inst), Merck (Inst), Merck KGaA (Inst), Novartis (Inst), Roche (Inst), and Servier (Inst). Ghassan K. Abou-Alfa: Research grants from Arcus, AstraZeneca, BioNtech, Bristol Myers Squibb, Celgene, Flatiron Health, Genentech/Roche, Genoscience Pharma, Incyte Corporation, Polaris Group, Puma Biotechnology, QED Therapeutics, Silenseed, Yiviva; and consultancy with Adicet Bio, Alnylam Pharmaceuticals, AstraZeneca, Autem Therapeutics, BeiGene, Berry Genomics, Boehringer Ingelheim, Celgene, Cend Therapeutics, CytomX, Eisai, Eli Lilly & Company, Exelixis, Flatiron Health, Genentech/Roche, Genoscience Pharma, Helios Pharmaceuticals, Helsinn Group, Incyte Corporation, Ipsen, Merck, Nerviano Medical Sciences, NewBridge Pharmaceuticals, Novartis, QED Therapeutics, Redhill, Rafael Pharmaceuticals, Servier, Silenseed, Sobi, Vector Pharma, and Yiviva.
Availability of data and material
Incyte Corporation (Wilmington, DE, USA) is committed to data sharing that advances science and medicine while protecting patient privacy. Qualified external scientific researchers may request anonymized datasets owned by Incyte for the purpose of conducting legitimate scientific research. Researchers may request anonymized datasets from any interventional study (except phase I studies) for which the product and indication have been approved on or after 1 January 2020 in at least one major market (e.g., United States, European Union, Japan). Data will be available for request after the primary publication or 2 years after the study has ended. Information on Incyte’s clinical trial data sharing policy and instructions for submitting clinical trial data requests are available at: https://www.incyte.com/Portals/0/Assets/Compliance%20and%20Transparency/clinical-trial-data-sharing.pdf?ver=2020-05-21-132838-960.
Ethics approval
The study, registered under ClinicalTrials.gov (NCT03144661), was performed in accordance with the International Conference for Harmonization Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable local regulations with approval from institutional review boards. The study protocol was reviewed and approved by the respective institutional review boards of the participating institutions (Online Supplementary Table 1).
Consent to participate
Written informed consent was obtained for all patients before performing study-related procedures.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
All authors were responsible for data collection, data integrity and analyses, and data interpretation, and reviewed and edited the manuscript. The corresponding author wrote the initial draft manuscript, had full access to all study data, and had final responsibility for the decision to submit for publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Harding, J.J., Jungels, C., Machiels, JP. et al. First-in-Human Study of INCB062079, a Fibroblast Growth Factor Receptor 4 Inhibitor, in Patients with Advanced Solid Tumors. Targ Oncol 18, 181–193 (2023). https://doi.org/10.1007/s11523-023-00948-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-023-00948-8