
Abstract
Sporadic desmoid-type fibromatosis is a rare, fibroblastic soft-tissue neoplasm with local aggressiveness but no metastatic potential. Aberrant Wnt/β-catenin signalling has been extensively linked to desmoid pathogenesis, although little is known about other molecular drivers and no established treatment approach exists. We aimed to summarise the current literature regarding the molecular pathogenesis of sporadic desmoid-type fibromatosis and to discuss the effects of both current and emerging novel therapies targeting these mechanisms. A literature search was conducted of MEDLINE® ALL and EMBASE databases for published studies (2000–August 2021) using keywords related to ‘fibromatosis aggressive’, ‘immunohistochemistry’, ‘polymerase chain reaction’ and ‘mutation’. Articles were included if they examined the role of proteins in sporadic or extra-abdominal human desmoid-type fibromatosis pathogenesis. Searching identified 1684 articles. Following duplicate removal and eligibility screening, 36 were identified. After a full-text screen, 22 were included in the final review. At least 47% of desmoid-type fibromatosis cases displayed aberrant β-catenin immunoreactivity amongst ten studies. Cyclin D1 overexpression occurred in at least 40% of cases across five studies. Six studies reported oestrogen receptor-β expression with a range of 7.4–90%. Three studies implicated matrix metalloproteinases, with one study demonstrating vascular endothelial growth factor overexpression. One study explored the positive relationship between cyclooxygenase-2 and platelet-derived growth factor receptor-β. Aberrant Wnt/β-catenin signalling is a well-established pathogenic driver that may be targeted via downstream modulation. Growth factor signalling is best appreciated through the clinical trial effects of multi-targeted tyrosine kinase inhibitors, whilst oestrogen receptor expression data may only offer a superficial insight into oestrogen signalling. Finally, the tumour microenvironment presents multiple potential novel therapeutic targets.
Plain Language Summary
Sporadic desmoid tumours are rare soft-tissue neoplasms that arise from connective tissues in the chest wall, head, neck and limbs. Whilst lacking metastatic potential, uncertainty surrounding their locally aggressive growth and unpredictable recurrence complicates treatment approaches. At the molecular level, alterations in the Wnt/β-catenin signalling pathway, a fundamental coordinator of cell growth and development, have been strongly linked to desmoid tumour development. Beyond this, however, little is known about other molecular drivers. In the case of progressive or life-threatening disease, complex treatment decisions are made regarding the use of surgery, radiotherapy or systemic treatment modalities. Of the targeted systemic therapies, a lack of comparative clinical studies further complicates medical treatment decision making as no definitive treatment approach exists. Therefore, this review aimed to summarise the literature regarding the molecular drivers of desmoid tumour pathogenesis and to discuss the current and emerging novel therapies targeting such mechanisms. Utilising findings from human desmoid tissue samples, we present the rationale for targeting downstream mediators of the central Wnt/β-catenin pathway and outline potential treatment targets in the tumour microenvironment. We also highlight the knowledge gained from clinical drug trials targeting desmoid growth factor signalling and present the potentially superficial insight provided by oestrogen receptor expression profiles on the role of oestrogen signalling in desmoid pathogenesis. In doing so, this work may assist in the eventual development of an evidence-based treatment approach for sporadic desmoid tumours.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Aberrant Wnt/β-catenin signalling is a well-established driver of desmoid tumour pathogenesis that may be effectively targeted via downstream blockade. |
The role of growth factors in desmoid tumour pathogenesis is best appreciated through the clinical trial success of tyrosine kinase inhibitor drugs targeting such factors. |
Oestrogen receptor expression data may only offer a superficial insight into oestrogen signalling mechanisms with clinical findings opposing anti-hormonal therapy, although further treatment opportunities exist within the tumour microenvironment. |
1 Introduction
Desmoid-type fibromatosis (DTF), also known as desmoid tumour or aggressive fibromatosis, is a rare soft-tissue neoplasm defined histologically by a monoclonal fibroblastic proliferation. These tumours arise in musculoaponeurotic structures and are characterised by locally infiltrative growth and a tendency towards local recurrence with no metastatic potential [1]. The estimated incidence of DTF lies between two and five cases per million people per year [2, 3].
Aetiological characteristics define two main groups: sporadic and familial DTF. Sporadic DTF comprises 85–90% of total cases that arise predominantly in extra-abdominal (E-AD) locations and have a slightly higher incidence following trauma, surgery, oral contraceptive use or within female individuals of reproductive age [2, 4,5,6,7,8,9]. In contrast, familial DTF represents an inheritable form associated with familial adenomatous polyposis (FAP). These tumours differ both clinically and pathologically from their sporadic counterparts because of their prevailing abdominal (AD) wall or intra-abdominal (I-AD) mesenteric and visceral locations and underlying APC gene mutations [2, 4, 5].
Desmoid pathogenesis has been associated with a number of signalling pathway aberrations. Of these, Wnt/β-catenin signalling has been extensively linked to desmoid pathogenesis (Fig. 1) [10,11,12,13,14]. In sporadic DTF, this is demonstrated by the vast majority of cases harbouring activating β-catenin gene (CTNNB1) mutations [15,16,17,18]. Beyond this signalling cascade, however, little is known about other molecular drivers implicated in desmoid pathogenesis and their interactions with Wnt/β-catenin signalling.

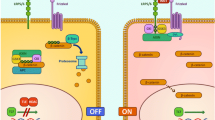
Wnt/β-catenin signalling pathway. The Wnt/β-catenin pathway coordinates cell proliferation, differentiation and fate during both embryogenesis and in normal adult tissues. a In the absence of a Wnt signal, cytoplasmic β-catenin that is not involved in cell-cell adhesion interacts with a degradation complex comprising axin, APC, GSK3 and CK1. Here, the sequential phosphorylation of β-catenin by CK1 and GSK3 marks it for ubiquitylation and degradation. This constant degradation prevents β-catenin from entering the nucleus and promoting the transcription of Wnt target genes. b The binding of Wnt to its frizzled receptor and LRP co-receptor leads to the recruitment of dishevelled. Together, this complex recruits the degradation complex to the cell membrane where LRP becomes phosphorylated by GSK3 then CK1. Axin then binds to the phosphorylated LRP, resulting in the disassembly of the degradation complex. Consequently, the stabilisation of β-catenin allows it to accumulate and translocate into the nucleus. Here, it binds to the TCF/LEF promotor region to stimulate the transcription of Wnt target genes including CCND1 (cyclin D1), MYC, PTGS2 (cyclooxygenase-2), MMP7, VEGF and WISP1. With deregulated, constitutive activation, the resultant protein products may drive tumourigenesis by enhancing proliferation, angiogenesis and invasiveness [10,11,12,13,14]. Created with BioRender.com
Owing to disease rarity, unpredictable clinical course and spontaneous regression rates, the current evidence base supports an “active surveillance” approach with close radiological monitoring in patients with stable non-critical disease. In the case of persistent progression or involvement of life-threatening anatomical sites lies the complex decision to engage active treatment options such as surgery, radiotherapy, chemotherapy or novel therapeutics [9, 19,20,21]. Particularly in the latter group, a lack of comparative clinical studies has prevented the creation of a definitive treatment approach for the implementation of systemic targeted therapies such as anti-hormonal therapy or tyrosine kinase inhibitors (TKIs) [21].
Improving our understanding of DTF growth and development would add greater precision to novel therapeutic decisions and may improve desmoid treatment outcomes. Therefore, this review aimed to summarise the current literature regarding the molecular pathogenesis of sporadic DTF and to discuss the effects of both current and emerging novel therapies targeting these mechanisms.
2 Methods
2.1 Search Strategy
A literature search was conducted on 7 August, 2021 using Ovid MEDLINE® ALL and EMBASE in consultation with a professional librarian. The search was limited to articles published after 1999 and an English language filter was applied. Keywords included ‘fibromatosis aggressive’, ‘immunohistochemistry’, ‘polymerase chain reaction’ and ‘mutation’ (see Tables 1, 2).
2.2 Article Eligibility and Study Selection
The titles and abstracts of records identified from database searching were assessed according to the eligibility criteria outlined in Table 3. Next, retrieved full-text articles were further assessed using the eligibility criteria listed in Table 3. Owing to the rare nature of desmoid tumours and the propensity for FAP-associated disease to occur in I-AD or AD locations [2, 4, 5], articles were excluded if they comprised > 15% of patients with FAP or included > 30% of AD or I-AD cases with no independent E-AD analysis (see Fig. 2).

Exclusion algorithm for retrieved articles using sporadic status and tumour location. E-AD extra-abdominal
2.3 Data Extraction
The first author, publication year, study type and methodological techniques were collected from all included papers. Clinical characteristics included the number of participants, number of primary and recurrent tumour samples, age, sex, sporadic status, tumour location and size. Tumour location was grouped into three broad categories. E-AD comprised the head and neck, pectoral and pelvic girdle, chest, upper and lower extremities, AD comprised the abdominal wall and I-AD comprised the abdominal cavity. Immunohistochemistry, DNA and mRNA sequencing data were retrieved for each studied protein.
3 Results
3.1 Systematic Literature Search
The search strategy yielded 1684 articles. Duplicates were then removed via computational software, leaving 1183 articles. The title and abstract were then screened, and a further 1147 articles were excluded. Of the 36 articles sought for retrieval, 22 articles were included in the final review (Fig. 3).

Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) search strategy flow diagram. FAP familial adenomatous polyposis
3.2 Study Design and Quality Assessment
All included studies were retrospective case series. Although some studies included control groups to assist with their deductions, these were not considered case-control studies as the features of these groups remained implicit and unclear [22]. The Joanna Briggs Institute critical appraisal tool for case series was utilised to determine each study’s risk of bias across nine domains (Fig. 4) [23]. ‘Overall’ judgement is described in Table 4. The domain concerning follow-up results was omitted as this was outside the scope of the review.

Joanna Briggs Institute critical appraisal for case-series via ROBINS-I [54]
3.3 Study Findings
Multiple proteins and associated signalling pathways have been linked to DTF pathogenesis (Table 5) with each pathway possessing potential therapeutic targets. Figure 5 outlines the key molecular drivers and associated pathophysiological domains identified by this review.

Key molecular drivers associated with desmoid tumour pathophysiology. a β-Catenin primarily coordinates cell proliferation, differentiation and fate, with its deregulated signalling being intrinsically linked to the development of several human cancers, such as skin, colon and breast cancer [13, 14]. Cyclin D1 and c-Myc signalling is commonly deregulated in tumourigenesis due to their promotional effects on cell proliferation by enhancing the G1 to S-phase transition of the cell cycle [34, 35]. b Cyclooxygenase-2 (COX2) is an inducible member of the cyclooxygenase family involved in multiple physiological purposes. In colorectal cancer, COX2 has been extensively implicated in promoting angiogenesis, invasion and proliferation through the upregulation of growth factors such as platelet-derived growth factor [37, 38]. The multi-functional transforming growth factor-beta (TGFβ) superfamily and related growth factors play complex and often opposing roles in cell proliferation, differentiation, regeneration and morphogenesis [41]. c Oestrogens are steroid hormones that promote growth, differentiation and reproduction throughout a range of human tissues. Their role in tumourigenesis has been extensively studied in breast cancer, where aberrant signalling drives proliferation, invasion and metastasis [44]. d Matrix metalloproteinases (MMPs) and related proteases play a pivotal role in cancer pathogenesis through their modulation of extracellular matrix, angiogenesis, cell migration and growth [48]. Vascular endothelial growth factor (VEGF) is a prominent angiogenic mediator whose expression is commonly upregulated in cancer tissue by various oncogenes, growth factors and hypoxia to sustain growth and invasion [52]. e The tumour suppressor genes RB1, CDKN2A and TP53 inhibit cell proliferation by arresting cells in the G1 phase of the cell cycle, with the latter protein being upregulated in response to DNA damage [53, 55]. f In its cell membrane function, β-catenin complexes with other proteins, such as α-catenin and N-cadherin, to mediate epithelial cell-cell adhesion and stability [57]. Created with BioRender.com. PDGFR platelet-derived growth factor receptor
3.3.1 Proliferation
3.3.1.1 β-Catenin
β-Catenin primarily coordinates cell proliferation, differentiation and fate, with its deregulated signalling being intrinsically linked to the development of several human cancers, such as skin, colon and breast cancer [13, 14]. β-Catenin demonstrated aberrant nuclear immunoreactivity in at least 47% of DTF cases [24,25,26,27,28,29,30,31,32,33]. DTF cases with a mutated CTNNB1 gene also demonstrated more frequent β-catenin nuclear expression (39/43, 90.7%) compared with cases with wild-type CTNNB1 (Fisher’s exact test, 17/27, 63.0%, p = 0.012) [28]. Saito et al. also demonstrated this difference with the mutated CTNNB1 group showing significantly higher β-catenin mRNA expression compared with the wild-type group (Mann–Whitney U test, p = 0.0036) [31]. DTF cases with abnormal β-catenin accumulation demonstrated a significantly higher proliferating cell nuclear antigen-labelling index than those with normal staining patterns (Fisher’s exact test, p = 0.007) [27]. Nuclear β-catenin immunoreactivity was significantly higher in desmoid tumours compared with both hypertrophic scar and normal fibrous tissue (Kruskal–Wallis test, p = 0.0003; Dunn’s post-tests, p < 0.01) [32]. These results are consistent with the current knowledge of Wnt/β-catenin’s role in DTF pathogenesis.
3.3.1.2 Cyclin D1 and c-Myc
Cyclin D1 and c-Myc signalling is commonly deregulated in tumourigenesis due to their promotional effects on cell proliferation by enhancing the G1 to S-phase transition of the cell cycle [34, 35]. Cyclin D1 was overexpressed in at least 40% of DTF cases [26, 27, 29, 36]. Two studies found a significant correlation between β-catenin nuclear expression and cyclin D1 overexpression (Fisher’s exact test, p = 0.029; Fisher’s exact test, p = 0.034, respectively) [27, 29]. Similarly, Jilong et al. found a significantly higher c-Myc expression in cases with abnormal β-catenin staining compared with normal cytomembrane staining (χ2 = 15.68, p = 0.0001) [26]. Furthermore, three studies demonstrated significantly higher CCND1 gene expression in the mutated CTNNB1 group compared with the wild-type group (χ2 test, p = 0.038; Mann–Whitney U test, p = 0.019; Mann–Whitney U test, p = 0.0120, respectively) [26, 29, 31]. Saito et al. and Santti et al. demonstrated significant cellular proliferative activity in DTF cases with cyclin D1 overexpression via a proliferating cell nuclear antigen-labelling index (Fisher’s exact test, p = 0.004) and Ki-67 (r = 0.40, p = 0.001), respectively [27, 36]. These findings suggest aberrant Wnt/β-catenin signalling may exert its proliferative effects through the overexpression of positive cell-cycle regulatory proteins.
3.3.2 Growth Factor Regulatory Signalling
3.3.2.1 Cyclooxygenase-2 (COX2) and Platelet-Derived Growth Factor Receptor (PDGFR)
COX2 is an inducible member of the cyclooxygenase family involved in multiple physiological purposes. In colorectal cancer, COX2 has been extensively implicated in promoting angiogenesis, invasion and proliferation through the upregulation of growth factors such as platelet-derived growth factor [37, 38]. Nuclear β-catenin did not correlate with COX2 expression (p = 0.034, p = 0.873) [32]. However, COX2 immunoreactivity was significantly higher in desmoid tumours compared with both hypertrophic scar and normal fibrous tissue (Kruskal–Wallis test, p < 0.0001; Dunn’s post-tests, p < 0.01) [32]. Signoroni et al. demonstrated 100% COX2, PDGFRα and PDGFRβ immunoreactivity and phosphorylation in their cohort of eight sporadic patients [39]. Cates et al. found 100% PDGFRβ expression in DTF samples (27/27) with a significantly higher immunoreactivity compared with normal fibrous tissue [40]. Albeit in a small cohort, COX2 induction may be responsible for the PDGFR expression validated by desmoid TKI trials, although this appeared to occur independently of β-catenin signalling.
3.3.2.2 Transforming Growth Factor-β (TGFβ) Superfamily and Other Growth Factors
The multifunctional TGFβ superfamily and related growth factors play complex and often opposing roles in cell proliferation, differentiation, regeneration and morphogenesis [41]. Two studies examined TFGβ signalling. Varghese et al. illustrated moderate-strong TGFβ and connective tissue growth factor (CTGF) immunoreactivity in 100% and 66.7% of DTF cases, respectively [33]. Mignemi et al. demonstrated phosphorylated SMAD2/3 immunoreactivity in 96% of their DTF cohort, which was significantly greater than both hypertrophic scar and normal fibrous tissue (Kruskal–Wallis test, p < 0.0001; Dunn’s post-hoc test, p < 0.001) [32]. Cates et al. found weak MET expression in 89% of DTF cases that differed significantly from normal fibrous tissue (Kruskal–Wallis test, p = 0.0005; Dunn’s post-hoc test, p < 0.001) [40]. Two studies evaluated epidermal growth factor receptor (EGFR) expression, which was detected in 11% of cases [30, 40]. Akt expression was detected in 56% of samples, which was significantly lower than the levels observed in hypertrophic scars (Kruskal–Wallis test, p = 0.0002; Dunn’s post-hoc test, p < 0.01). No study demonstrated positive expression for c-Kit [30, 42, 43] or human epidermal growth factor receptor 2 (HER2) [30, 43]. TGFβ and related growth factors appeared to demonstrate aberrant expression in DTF, although the direct pathological consequence of this finding remains unclear.
3.3.3 Oestrogen-Related Pathway
Oestrogens are steroid hormones that promote growth, differentiation and reproduction throughout a range of human tissues. Their role in tumourigenesis has been extensively studied in breast cancer, where aberrant signalling drives proliferation, invasion and metastasis [44]. Six studies evaluated the role of sex steroids in desmoid tumour pathogenesis [36, 42, 43, 45,46,47]. Oestrogen receptor-β (ERβ) expression was demonstrated in all studies and ranged from 7.4% to 90% [36, 42, 43, 45,46,47], whilst only Ishizuka et al. demonstrated oestrogen receptor-α (ERα) expression in two patients [46]. ERβ significantly correlated with the expression of cyclin A (r = 0.34, p = 0.004), cyclin D1 (r = 0.34, p = 0.004) and Ki-67 (r = 0.35, p = 0.003) [36]. Ishizuka et al. also demonstrated positive progesterone receptor A and B expression in 25.9% and 33.3% of DTF cases respectively [46], whilst the remaining studies were negative for progesterone receptor expression [42, 43, 47]. In addition to sex steroid receptor analysis, Brautigam et al. found poly(ADP-ribose) polymerase 1 (PARP1) expression in 98.3% of DTF cases, which negatively correlated with Ki-67 expression (Spearman–Rho test, − 0.375, p = 0.041) [47]. In the context of widespread ERβ expression and its positive correlation with proliferation markers, these findings support a proliferative role for oestrogen in desmoid pathogenesis.
3.3.4 Tumour Microenvironment
3.3.4.1 Invasion
Matrix metalloproteinases (MMPs) and related proteases play a pivotal role in cancer pathogenesis through their modulation of the extracellular matrix, angiogenesis, cell migration and growth [48]. Four studies evaluated the expression of MMPs [29, 49,50,51]. β-Catenin nuclear expression significantly correlated with MMP7 overexpression (Fisher’s exact test, p < 0.01), and mutated CTNNB1 also significantly increased MMP7 mRNA expression compared with wild-type CTNNB1 (Mann–Whitney U test, p = 0.0018) [29]. Colombo et al. demonstrated an increased MMP2 staining intensity in tumours with CTNNB1 mutations compared with wild-type cases (Fisher’s exact test, p = 0.0438) [49]. High MMP2 and EMMPRIN mRNA expression was seen in 57.1% and 42.9% of DTF respectively, but their expression did not differ significantly from benign fibrous tumours (Fisher’s exact test, p > 0.05) [50]. Two studies evaluated the expression of ADAM12 [49, 51]. ADAM12 expression was observed in 195 DTF cases, and its expression positively correlated with chromosome density (r = 0.30, p < 0.001) [49, 51]. Misemer et al. also demonstrated a significant correlation between chromosome density and Wnt inducible signalling pathway protein 1 (WISP1) (r = 0.27, p < 0.001) as well as fibroblast activation protein alpha (FAP-alpha) expression (r = 0.44, p < 0.001) [51]. MMPs and related proteases were found to be over-expressed in DTF samples, with their expression further enhanced by the presence of aberrant β-catenin signalling.
3.3.4.2 Angiogenesis
Vascular endothelial growth factor (VEGF) is a prominent angiogenic mediator whose expression is commonly upregulated in cancer tissue by various oncogenes, growth factors and hypoxia to sustain growth and invasion [52]. Matono et al. demonstrated a significant correlation between β-catenin nuclear immunoreactivity and VEGF overexpression (Fisher’s exact test, p = 0.04). Mean microvessel density was also significantly higher in recurrent (13.97 mm2) compared with primary tumours (9.56 mm2, Fisher’s exact test, p = 0.02). VEGF-positive samples showed a trend towards a higher microvessel density, but this did not reach statistical significance (VEGF-positive, 10.62 mm2; VEGF-negative, 9.55 mm2; Fisher’s exact test, p = 0.84) [24]. Furthermore, Colombo et al. confirmed the overexpression of midkine seen on gene expression profiling with 46% of DTF samples illustrating immunoreactivity [49]. These results highlight VEGF as a key angiogenic mediator in DTF pathogenesis with its expression appearing to occur in a β-catenin-dependent manner.
3.3.5 Cell-Cycle Regulatory Proteins
The tumour suppressor genes RB1, CDKN2A and TP53 inhibit cell proliferation by arresting cells in the G1 phase of the cell cycle, with the latter protein being upregulated in response to DNA damage [53, 55]. Stalinska et al. showed normal pRb and p16 expression in 94.4% and 50% of E-AD cases, respectively. Heterogeneous or low expression of pRb and p16 was seen in 5.56% and 50% of E-AD cases, respectively [56]. Gebert et al. found p53 expression in 32% of cases, which significantly correlated with β-catenin expression (χ2 test, p < 0.05) [30]. The normal expression profiles of both pRb and p16 suggest an intact G1 cell-cycle regulatory function, whilst the increased p53 expression suggests a degree of aberrant cell-cycle progression in the context of β-catenin expression.
3.3.6 Cell–Cell Adhesion
In its cell membrane function, β-catenin complexes with other proteins, such as α-catenin and N-cadherin, to mediate epithelial cell-cell adhesion and stability [57]. Ferenc et al. reported a lack of α-catenin expression in 34% of their sporadic DTF cases. E-AD cases demonstrated a significantly higher mean cellular α-catenin immunoreactivity at 61.5% compared with 42.3% in AD cases (Student’s t test, p = 0.0165). N-cadherin expression was positive in 23% of all DTF cases. Nuclear β-catenin expression did not significantly correlate with α-catenin or N-cadherin expression (Pearson’s correlation, p > 0.05) [25]. This study indicates a possible progressive tumour phenotype characterised by the loss of cell-cell adhesion markers, although the findings did not reach statistical significance.
4 Discussion
This review critically examined the current literature regarding the molecular pathogenesis of sporadic DTF in human tumour samples and utilised its findings to explore current and emerging novel therapies. Twenty-two articles exploring the molecular pathogenesis were included. Within these, Wnt/β-catenin pathway aberrations were extensively studied. Oestrogen, growth factor regulatory signalling and tumourigenic microenvironment changes were also implicated.
The role of aberrant Wnt/β-catenin signalling in desmoid pathogenesis is well established [15,16,17,18]. Accordingly, this review found aberrant β-catenin expression in almost half of DTF cases within each study [24,25,26,27,28,29,30,31,32,33]. Mutated β-catenin also enhanced its pathologic role compared with its wild-type counterpart, producing greater overexpression of downstream target genes such as cyclin D1 [26, 27, 29, 31]. DTF commonly harbors CTNNB1 mutations in position T41 and S45, which enhance β-catenin’s stability by decreasing phosphorylation-guided degradation (Fig. 6) [15, 58]. Additionally, Crago et al.’s next-generation sequencing study suggests a more pervasive role for mutated β-catenin with Wnt/β-catenin-activating mutations found in 95% of samples compared with 86% with conventional Sanger sequencing (n = 117) [15]. Although Wnt blockade presents a desirable therapeutic target, adverse effects on immune function and gastrointestinal homeostasis have precluded the development of prospective sporadic DTF trials [14, 59]. In solid tumours, however, the decoy Wnt ligand receptor ipafricept (OMP-54F28) was evaluated in a phase I clinical trial, with two patients with DTF experiencing stable disease beyond 6 months [60]. In light of the undesirable effects of Wnt blockade, perhaps targeting more distal branches of the pathway may be of greater utility. In colorectal cancer, the demonstration of Wnt/β-catenin signalling crosstalk with the Notch pathway has prompted the use of targeted therapies against this interaction [61,62,63]. Most notably, the γ-secretase inhibitor nirogacestat (PF-03084014), a small-molecule drug that prevents activation of the Notch intracellular domain, was evaluated in a phase II clinical trial comprising 17 patients with actively progressive DTF disease. Here, 29% experienced a partial response after a median of 32 cycles with a further 65% achieving stable disease [64]. Following its success, the phase III DeFi trial is currently in progress evaluating nirogacestat in a similar cohort of adult patients with DTF, with the primary completion date reached in December 2021 [65]. Additionally, as a cell-cycle promoter, cyclin D1 blockade may also negatively augment desmoid growth by inhibiting the G1 to S phase transition of the cell cycle [34]. Palbociclib, an inhibitor of the cyclin D1 activating protein Cdk4/6, demonstrates an overall survival benefit in a randomised trial involving patients with advanced breast cancer [66]. With cyclin D1 emerging as an overactive protein, its inhibition may present an attractive therapeutic target.

Aberrant Wnt/β-catenin signalling with mutated CTNNB1. T41A and S45F represent the two most common substitution mutations harboured by sporadic desmoid-type fibromatosis. These amino acid substitutions prevent β-catenin’s phosphorylation by GSK3 and CK1. Consequently, the mutated β-catenin is not marked for degradation, allowing it to accumulate and translocate into the nucleus where it promotes the unregulated transcription of specific target genes. The resultant protein products drive tumourigenesis by enhancing proliferation, angiogenesis and invasiveness [15, 58]. Created with BioRender.com. COX2 cyclooxygenase-2, MMP matrix metalloproteinase, S45F serine to phenylalanine substitution in codon 45, T41A threonine to alanine substitution in codon 41, VEGF vascular endothelial growth factor, WISP1 Wnt inducible signalling pathway protein 1
Targeted growth factor inhibitory therapy is a promising treatment modality in DTF management. Historically, the benefits of TKIs on small DTF cohorts were rationalised by the potential expression of c-Kit and PDGFR [67]. Since then, the absence of c-Kit staining has negated this view [43, 68]. Accordingly, this review reported no c-Kit expression [30, 42, 43]. Consequently, the use of TKIs is now rationalised by the demonstration of PDGFR expression. In this review, only one study observed the expression and phosphorylation of PDGFRβ in a small DTF cohort with COX2 overexpression (n = 8 sporadic), with a larger cohort comprising 27 sporadic DTF samples demonstrating a significantly higher PDGFRβ expression compared with normal fibrous tissue [39, 40]. Evidently, because of the limited focus on human DTF samples, our findings offer an incomplete understanding on the role of growth factors in desmoid pathogenesis with a much greater appreciation coming from the expanding evaluation of TKI therapy. The first TKI trialled in DTF, imatinib, is a multi-targeted TKI that primarily inhibits c-Kit and PDGFRβ [69]. In a phase II trial comprising 40 patients with unresectable and progressive DTF, the primary endpoint of non-progressive disease at 3 months is 91%, with a 2-year progression-free survival rate of 55% [70]. In a similar DTF cohort, the phase III double-blind trial testing the multi-targeted TKI sorafenib produces a 2-year progression-free survival rate of 81% compared with 36% in the placebo group [71]. Furthermore, in the phase II DESMOPAZ trial, the antiangiogenic TKI pazopanib prevents disease progression over 6 months in 84% compared with 45% in patients receiving methotrexate-vinblastine combination chemotherapy [72]. In addition to the mechanism of imatinib, both sorafenib and pazopanib also inhibit VEGFR2/3 [69], suggesting the angiogenic component of DTF pathogenesis may prove a more attractive therapeutic target than PDGFR alone.
A growing body of retrospective evidence implicates oestrogen in desmoid pathogenesis. Clinically, DTF arises more commonly in female individuals of reproductive age, with accelerated tumour growth observed during pregnancy and with oral contraceptive use [5,6,7,8,9]. Supporting these observations at the molecular level, this review identified ERβ expression in all respective studies, albeit within a broad range of expression values and a low proportion of positively stained cells [36, 42, 43, 45,46,47]. Furthermore, a positive correlation was also demonstrated between ERβ expression, cyclin D1 and proliferation markers [36]. This finding further supports the emerging anabolic role of ERβ signalling that has previously been described in promoting the regeneration of injured skeletal muscle tissue [73, 74]. This, in conjunction with its opposing tumour-suppressor effects in breast cancer [75], suggests a tissue-specific proliferative role in mesenchymal tissues. Although these histological data provide a potential rationale for oestrogen blockage in DTF treatment, studies evaluating its effect have produced conflicting results. Supporting its use, a systematic review of anti-oestrogen therapy found tamoxifen to produce an overall response rate of 58% (n = 22/38) from a cohort comprising 47.7% sporadic DTF [76]. In the only prospective evaluation of anti-hormonal therapy, however, the combination of tamoxifen and sulindac in a phase II trial produces an overall response rate and 2-year progression-free survival rate of 8% and 36%, respectively, in a paediatric cohort of 59 patients with DTF [77]. In light of such results, the use of anti-hormonal therapy and non-steroidal anti-inflammatory drugs is not endorsed by The Desmoid Tumour Working Group’s latest consensus-based treatment guidelines [21]. Therefore, this review may only provide a superficial insight into the role of oestrogen receptor signalling in desmoid pathogenesis. Whilst we did not find anything to contradict the use of anti-hormonal therapy, this review was limited in its search to human DTF samples only, and there was no exploration of second messenger co-activators or repressors that may explain the negligible effects of therapeutic oestrogen blockade.
The tumour microenvironment is emerging as a key player in sustaining desmoid cell growth and longevity. This review demonstrated positive MMP7, MMP2 and ADAM12 expression, with mutated CTNNB1 significantly increasing both MMP7 gene expression and MMP2 immunoreactivity [29, 49,50,51]. Surprisingly, MMP2 expression did not differ significantly from benign fibrous tumours [50]. In light of these findings, it remains unclear whether this MMP overexpression suggests an augmentation of physiological function or a hallmark feature of tumourigenesis, as associations with defining features such as invasion, aberrant growth factor signalling and angiogenesis [48] were not explored. Supporting the pathologic role, the MMP inhibitor ilimostat decreases DTF cell invasion in human cell cultures [78] as well as DTF cell invasion and motility in Apc+/Apc1638N mice [79]. Historically, however, experimental MMP inhibitor therapy has translated poorly to clinical studies because of their previously unrecognised anti-tumour role [80]. Furthermore, overexpression of the key angiogenic mediator VEGF significantly correlated with β-catenin nuclear reactivity and demonstrated a higher microvessel density compared with VEGF-negative tumours [24]. Similarly, Meazza et al. also identified the Q472H VEGFR2 polymorphism in 56% and 40% of their paediatric and adult patients, respectively [81]. These findings, together with previously described efficacy of both sorafenib and pazopanib, suggest a proangiogenic phenotype that may benefit from targeted therapy. Exploring this vascular inhibition, the antiangiogenic protein endostatin directly induces cell death in vitro on primary FAP-related DTF cells [82], although effects on sporadic DTF cells are yet to be explored.
4.1 Limitations
The inclusion of retrospective case series limited this review because of their high level of bias [22]. Eight studies also potentially utilised overlapping patient cohorts as suggested by shared authorship, inclusion periods and specimen archives [24, 25, 27, 29, 31, 32, 40, 56]. Furthermore, key clinical information such as age, sex, tumour size and sporadic status was omitted throughout included papers. Consequently, patients with FAP were inadvertently included in this review. Included studies also utilised varied cut-offs and staining patterns to define protein overexpression, leading to marked result heterogeneity. There were also a number of limitations to this review’s methodology. Only articles written in the English language and published after 1999 were included. The elucidation of DTF pathogenesis was also restricted by a number of factors, such as the exclusion of animal and cell culture studies, patient follow-up and treatment effects on molecular markers.
5 Conclusions
The presence of aberrant Wnt/β-catenin signalling in sporadic DTF pathogenesis is well established and may be effectively targeted via downstream augmentation. This review also elucidated the tumour microenvironment’s emerging role in desmoid development with preliminary evidence favouring angiogenic antagonism. This study is the first of its type to systematically review the molecular pathogenesis of human sporadic DTF in the era of targeted therapies. Future work may wish to further evaluate the additional signalling pathways implicated in DTF pathogenesis and the mechanisms of its associated novel therapies.
References
Fletcher CDM. WHO classification of tumours of soft tissue and bone. World Health Organization classification of tumours. Geneva: International Agency for Research on Cancer; 2013.
Nieuwenhuis MH, Casparie M, Mathus-Vliegen LM, Dekkers OM, Hogendoorn PC, Vasen HF. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer. 2011;129(1):256–61. https://doi.org/10.1002/ijc.25664.
van Broekhoven DL, Grünhagen DJ, den Bakker MA, van Dalen T, Verhoef C. Time trends in the incidence and treatment of extra-abdominal and abdominal aggressive fibromatosis: a population-based study. Ann Surg Oncol. 2015;22(9):2817–23. https://doi.org/10.1245/s10434-015-4632-y.
Gurbuz AK, Giardiello FM, Petersen GM, Krush AJ, Offerhaus GJ, Booker SV, et al. Desmoid tumours in familial adenomatous polyposis. Gut. 1994;35(3):377–81. https://doi.org/10.1136/gut.35.3.377.
Koskenvuo L, Ristimäki A, Lepistö A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J Surg Oncol. 2017;116(6):716–21. https://doi.org/10.1002/jso.24699.
Fiore M, Rimareix F, Mariani L, Domont J, Collini P, Le Péchoux C, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol. 2009;16(9):2587–93. https://doi.org/10.1245/s10434-009-0586-2.
Fiore M, MacNeill A, Gronchi A, Colombo C. Desmoid-type fibromatosis: evolving treatment standards. Surg Oncol Clin N Am. 2016;25(4):803–26. https://doi.org/10.1016/j.soc.2016.05.010.
Martínez Trufero J, Pajares Bernad I, Torres Ramón I, Hernando Cubero J, Pazo CR. Desmoid-type fibromatosis: who, when, and how to treat. Curr Treat Options Oncol. 2017;18(5):29. https://doi.org/10.1007/s11864-017-0474-0.
Kasper B, Baumgarten C, Bonvalot S, Haas R, Haller F, Hohenberger P, et al. Management of sporadic desmoid-type fibromatosis: a European consensus approach based on patients’ and professionals’ expertise: a sarcoma patients EuroNet and European Organisation for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group initiative. Eur J Cancer. 2015;51(2):127–36. https://doi.org/10.1016/j.ejca.2014.11.005.
Lecarpentier Y, Schussler O, Hébert J-L, Vallée A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front Oncol. 2019;9:1248. https://doi.org/10.3389/fonc.2019.01248.
Olsen JJ, Pohl S, Deshmukh A, Visweswaran M, Ward NC, Arfuso F, et al. The role of Wnt signalling in angiogenesis. Clin Biochem Rev. 2017;38(3):131–42.
Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ. WISP-1 is a Wnt-1- and beta-catenin-responsive oncogene. Genes Dev. 2000;14(5):585–95.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. https://doi.org/10.1016/j.devcel.2009.06.016.
Enzo MV, Rastrelli M, Rossi CR, Hladnik U, Segat D. The Wnt/β-catenin pathway in human fibrotic-like diseases and its eligibility as a therapeutic target. Mol Cell Ther. 2015;3:1. https://doi.org/10.1186/s40591-015-0038-2.
Crago AM, Chmielecki J, Rosenberg M, O’Connor R, Byrne C, Wilder FG, et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer. 2015;54(10):606–15. https://doi.org/10.1002/gcc.22272.
Aitken SJ, Presneau N, Kalimuthu S, Dileo P, Berisha F, Tirabosco R, et al. Next-generation sequencing is highly sensitive for the detection of beta-catenin mutations in desmoid-type fibromatoses. Virchows Arch. 2015;467(2):203–10. https://doi.org/10.1007/s00428-015-1765-0.
Trautmann M, Rehkamper J, Gevensleben H, Becker J, Wardelmann E, Hartmann W, et al. Novel pathogenic alterations in pediatric and adult desmoid-type fibromatosis: a systematic analysis of 204 cases. Sci. 2020;10(1):3368. https://doi.org/10.1038/s41598-020-60237-6.
Tejpar S, Nollet F, Li C, Wunder JS, Michils G, dal Cin P, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene. 1999;18(47):6615–20. https://doi.org/10.1038/sj.onc.1203041.
Penel N, Coindre JM, Bonvalot S, Italiano A, Neuville A, Le Cesne A, et al. Management of desmoid tumours: a nationwide survey of labelled reference centre networks in France. Eur J Cancer. 2016;58:90–6. https://doi.org/10.1016/j.ejca.2016.02.008.
Penel N, Le Cesne A, Bonvalot S, Giraud A, Bompas E, Rios M, et al. Surgical versus non-surgical approach in primary desmoid-type fibromatosis patients: a nationwide prospective cohort from the French Sarcoma Group. Eur J Cancer. 2017;83:125–31. https://doi.org/10.1016/j.ejca.2017.06.017.
Desmoid Tumor Working G. The management of desmoid tumours: a joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer. 2020;127:96–107. doi: https://doi.org/10.1016/j.ejca.2019.11.013.
Guyatt GH, Oxman AD, Vist G, Kunz R, Brozek J, Alonso-Coello P, et al. GRADE guidelines: 4. Rating the quality of evidence: study limitations (risk of bias). J Clin Epidemiol. 2011;64(4):407–15. https://doi.org/10.1016/j.jclinepi.2010.07.017.
JBI. Checklist for case series. 2020. https://jbi.global/critical-appraisal-tools. Accessed 27 Mar 2022.
Matono H, Tamiya S, Yokoyama R, Saito T, Iwamoto Y, Tsuneyoshi M, et al. Abnormalities of the Wnt/beta-catenin signalling pathway induce tumour progression in sporadic desmoid tumours: correlation between beta-catenin widespread nuclear expression and VEGF overexpression. Histopathology. 2011;59(3):368–75. https://doi.org/10.1111/j.1365-2559.2011.03945.x.
Ferenc T, Wronski JW, Kopczynski J, Kulig A, Sidor M, Stalinska L, et al. Analysis of APC, alpha-, beta-catenins, and N-cadherin protein expression in aggressive fibromatosis (desmoid tumor). Pathol Res Pract. 2009;205(5):311–24. https://doi.org/10.1016/j.prp.2008.11.002.
Jilong Y, Jian W, Xiaoyan Z, Xiaoqiu L, Xiongzeng Z. Analysis of APC/beta-catenin genes mutations and Wnt signalling pathway in desmoid-type fibromatosis. Pathology. 2007;39(3):319–25. https://doi.org/10.1080/00313020701329823.
Saito T, Oda Y, Tanaka K, Matsuda S, Tamiya S, Iwamoto Y, et al. beta-catenin nuclear expression correlates with cyclin D1 overexpression in sporadic desmoid tumours. J Pathol. 2001;195(2):222–8. https://doi.org/10.1002/path.942.
An J, Woo HY, Lee Y, Kim HS, Jeong J, Kim SK. Clinicopathological features of 70 desmoid-type fibromatoses confirmed by beta-catenin immunohistochemical staining and CTNNB1 mutation analysis. PLoS ONE. 2021;16(4): e0250619. https://doi.org/10.1371/journal.pone.0250619.
Matono H, Oda Y, Nakamori M, Tamiya S, Yamamoto H, Yokoyama R, et al. Correlation between beta-catenin widespread nuclear expression and matrix metalloproteinase-7 overexpression in sporadic desmoid tumors. Hum Pathol. 2008;39(12):1802–8. https://doi.org/10.1016/j.humpath.2008.05.005.
Gebert C, Hardes J, Kersting C, August C, Supper H, Winkelmann W, et al. Expression of beta-catenin and p53 are prognostic factors in deep aggressive fibromatosis. Histopathology. 2007;50(4):491–7. https://doi.org/10.1111/j.1365-2559.2007.02619.x.
Saito T, Oda Y, Kawaguchi K, Tanaka K, Matsuda S, Tamiya S, et al. Possible association between higher beta-catenin mRNA expression and mutated beta-catenin in sporadic desmoid tumors: real-time semiquantitative assay by TaqMan polymerase chain reaction. Lab Investig. 2002;82(1):97–103.
Mignemi NA, Itani DM, Fasig JH, Keedy VL, Hande KR, Whited BW, et al. Signal transduction pathway analysis in desmoid-type fibromatosis: transforming growth factor-beta, COX2 and sex steroid receptors. Cancer Sci. 2012;103(12):2173–80. https://doi.org/10.1111/cas.12037.
Varghese S, Braggio DA, Gillespie J, Toland AE, Pollock R, Mayerson J, et al. TGF-beta and CTGF are mitogenic output mediators of Wnt/beta-catenin signaling in desmoid fibromatosis. Appl Immunohistochem Molecul Morphol. 2017;25(8):559–65. https://doi.org/10.1097/PAI.0000000000000340.
Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14(8):518–28. https://doi.org/10.1038/nrm3629.
García-Gutiérrez L, Bretones G, Molina E, Arechaga I, Symonds C, Acosta JC, et al. Myc stimulates cell cycle progression through the activation of Cdk1 and phosphorylation of p27. Sci Rep. 2019;9(1):18693. https://doi.org/10.1038/s41598-019-54917-1.
Santti K, Ihalainen H, Ronty M, Karlsson C, Haglund C, Sampo M, et al. Estrogen receptor beta expression correlates with proliferation in desmoid tumors. J Surg Oncol. 2019;119(7):873–9. https://doi.org/10.1002/jso.25407.
Dempke W, Rie C, Grothey A, Schmoll HJ. Cyclooxygenase-2: a novel target for cancer chemotherapy? J Cancer Res Clin Oncol. 2001;127(7):411–7. https://doi.org/10.1007/s004320000225.
Kundu JK, Choi KY, Surh YJ. beta-Catenin-mediated signaling: a novel molecular target for chemoprevention with anti-inflammatory substances. Biochim Biophys Acta. 2006;1765(1):14–24. https://doi.org/10.1016/j.bbcan.2005.08.006.
Signoroni S, Frattini M, Negri T, Pastore E, Tamborini E, Casieri P, et al. Cyclooxygenase-2 and platelet-derived growth factor receptors as potential targets in treating aggressive fibromatosis. Clin Cancer Res. 2007;13(17):5034–40.
Cates JM, Black JO, Itani DM, Fasig JH, Keedy VL, Hande KR, et al. Signal transduction pathway analysis in fibromatosis: receptor and nonreceptor tyrosine kinases. Hum Pathol. 2012;43(10):1711–8. https://doi.org/10.1016/j.humpath.2011.12.021.
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30. https://doi.org/10.1038/nrm3434.
Santos GA, Cunha IW, Rocha RM, Mello CA, Guimaraes GC, Fregnani JH, et al. Evaluation of estrogen receptor alpha, estrogen receptor beta, progesterone receptor, and cKIT expression in desmoids tumors and their role in determining treatment options. Biosci Trends. 2010;4(1):25–30.
Leithner A, Gapp M, Radl R, Pascher A, Krippl P, Leithner K, et al. Immunohistochemical analysis of desmoid tumours. J Clin Pathol. 2005;58(11):1152–6.
Deyrup AT, Tretiakova M, Montag AG. Estrogen receptor-beta expression in extraabdominal fibromatoses: an analysis of 40 cases. Cancer. 2006;106(1):208–13. https://doi.org/10.1002/cncr.21553.
Ishizuka M, Hatori M, Dohi O, Suzuki T, Miki Y, Tazawa C, et al. Expression profiles of sex steroid receptors in desmoid tumors. Tohoku J Exp Med. 2006;210(3):189–98. https://doi.org/10.1620/tjem.210.189.
Brautigam K, Lindner J, Budczies J, Pahl S, Kunitz A, Melcher I, et al. PARP-1 expression as a prognostic factor in desmoid-type fibromatosis. Ann Diagn Pathol. 2020;44: 151442. https://doi.org/10.1016/j.anndiagpath.2019.151442.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. https://doi.org/10.1016/j.cell.2010.03.015.
Colombo C, Creighton CJ, Ghadimi MP, Bolshakov S, Warneke CL, Zhang Y, et al. Increased midkine expression correlates with desmoid tumour recurrence: a potential biomarker and therapeutic target. J Pathol. 2011;225(4):574–82. https://doi.org/10.1002/path.2951.
Ahlen J, Enberg U, Larsson C, Larsson O, Frisk T, Brosjo O, et al. Malignant fibrous histiocytoma, aggressive fibromatosis and benign fibrous tumors wxpress mRNA for the metalloproteinase inducer EMMPRIN and the metalloproteinases MMP-2 and MT1-MMP. Sarcoma. 2001;5(3):143–9. https://doi.org/10.1080/13577140120048601.
Misemer BS, Skubitz APN, Carlos Manivel J, Schmechel SC, Cheng EY, Henriksen JC, et al. Expression of FAP, ADAM12, WISP1, and SOX11 is heterogeneous in aggressive fibromatosis and spatially relates to the histologic features of tumor activity. Cancer Med. 2014;3(1):81–90. https://doi.org/10.1002/cam4.160.
Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl. 3):4–10. https://doi.org/10.1159/000088478.
Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. 2021;11(5):1913–35.
Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18(53):7644–55. https://doi.org/10.1038/sj.onc.1203015.
Lipovka Y, Konhilas JP. The complex nature of oestrogen signalling in breast cancer: enemy or ally? 2016. Biosci Rep. https://doi.org/10.1042/bsr20160017.
Stalinska L, Turant M, Tosik D, Sygut J, Kulig A, Kopczynski J, et al. Analysis of pRb, p16INK4A proteins and proliferating antigens: PCNA, Ki-67 and MCM5 expression in aggressive fibromatosis (desmoid tumor). Histol Histopathol. 2009;24(3):299–308.
Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36(Pt 2):149–55. https://doi.org/10.1042/bst0360149.
Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–99. https://doi.org/10.1016/j.cell.2017.05.016.
Timbergen MJM, Smits R, Grunhagen DJ, Verhoef C, Sleijfer S, Wiemer EAC. Activated signaling pathways and targeted therapies in desmoid-type fibromatosis: a literature review. Front Oncol. 2019;9:397. https://doi.org/10.3389/fonc.2019.00397.
Jimeno A, Gordon M, Chugh R, Messersmith W, Mendelson D, Dupont J, et al. A first-in-Human phase I study of the anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(24):7490–7. https://doi.org/10.1158/1078-0432.Ccr-17-2157.
Peignon G, Durand A, Cacheux W, Ayrault O, Terris B, Laurent-Puig P, et al. Complex interplay between β-catenin signalling and Notch effectors in intestinal tumorigenesis. Gut. 2011;60(2):166–76. https://doi.org/10.1136/gut.2009.204719.
Rodilla V, Villanueva A, Obrador-Hevia A, Robert-Moreno A, Fernández-Majada V, Grilli A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci USA. 2009;106(15):6315–20. https://doi.org/10.1073/pnas.0813221106.
Ungerbäck J, Elander N, Grünberg J, Sigvardsson M, Söderkvist P. The Notch-2 gene is regulated by Wnt signaling in cultured colorectal cancer cells. PLoS ONE. 2011;6(3): e17957. https://doi.org/10.1371/journal.pone.0017957.
Kummar S, O’Sullivan Coyne G, Do KT, Turkbey B, Meltzer PS, Polley E, et al. Clinical activity of the γ-secretase inhibitor PF-03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol. 2017;35(14):1561–9. https://doi.org/10.1200/jco.2016.71.1994.
SpringWorksTherapeutics. Nirogacestat for adults with desmoid tumor/aggressive fibromatosis (DT/AF) (DeFi). ClinicalTrials.gov. 2018. https://clinicaltrials.gov/ct2/show/NCT03785964?id=NCT03785964&draw=2&rank=1&load=cart. Accessed 27 Mar 2021.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926–36. https://doi.org/10.1056/NEJMoa1810527.
Mace J, Sybil Biermann J, Sondak V, McGinn C, Hayes C, Thomas D, et al. Response of extraabdominal desmoid tumors to therapy with imatinib mesylate. Cancer. 2002;95(11):2373–9. https://doi.org/10.1002/cncr.11029.
Hornick JL, Fletcher CD. Validating immunohistochemical staining for KIT (CD117). Am J Clin Pathol. 2003;119(3):325–7. https://doi.org/10.1309/ej3ry45qcypukqg4.
Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J Clin Oncol. 2011;2(2):80–93. https://doi.org/10.5306/wjco.v2.i2.80.
Penel N, Le Cesne A, Bui BN, Perol D, Brain EG, Ray-Coquard I, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol. 2011;22(2):452–7. https://doi.org/10.1093/annonc/mdq341.
Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. 2018;379(25):2417–28. https://doi.org/10.1056/NEJMoa1805052.
Toulmonde M, Pulido M, Ray-Coquard I, Andre T, Isambert N, Chevreau C, et al. Pazopanib or methotrexate-vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): a non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet Oncol. 2019;20(9):1263–72. https://doi.org/10.1016/s1470-2045(19)30276-1.
Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids. 2014;90:13–29. https://doi.org/10.1016/j.steroids.2014.06.012.
Velders M, Schleipen B, Fritzemeier KH, Zierau O, Diel P. Selective estrogen receptor-β activation stimulates skeletal muscle growth and regeneration. FASESB J. 2012;26(5):1909–20. https://doi.org/10.1096/fj.11-194779.
Zhou Y, Liu X. The role of estrogen receptor beta in breast cancer. Biomark Res. 2020;8(1):39. https://doi.org/10.1186/s40364-020-00223-2.
Bocale D, Rotelli MT, Cavallini A, Altomare DF. Anti-oestrogen therapy in the treatment of desmoid tumours: a systematic review. Colorectal Dis. 2011;13(12):e388–95. https://doi.org/10.1111/j.1463-1318.2011.02758.x.
Skapek SX, Anderson JR, Hill DA, Henry D, Spunt SL, Meyer W, et al. Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a Children’s Oncology Group (COG) phase II study. Pediatr Blood Cancer. 2013;60(7):1108–12. https://doi.org/10.1002/pbc.24457.
Denys H, De Wever O, Nusgens B, Kong Y, Sciot R, Le AT, et al. Invasion and MMP expression profile in desmoid tumours. Br J Cancer. 2004;90(7):1443–9. https://doi.org/10.1038/sj.bjc.6601661.
Kong Y, Poon R, Nadesan P, Di Muccio T, Fodde R, Khokha R, et al. Matrix metalloproteinase activity modulates tumor size, cell motility, and cell invasiveness in murine aggressive fibromatosis. Cancer Res. 2004;64(16):5795–803. https://doi.org/10.1158/0008-5472.Can-03-3112.
Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. 2018;17(6):1147–55. https://doi.org/10.1158/1535-7163.Mct-17-0646.
Meazza C, Belfiore A, Busico A, Settanni G, Paielli N, Cesana L, et al. AKT1 and BRAF mutations in pediatric aggressive fibromatosis. Cancer Med. 2016;5(6):1204–13. https://doi.org/10.1002/cam4.669.
Martinico SC, Jezzard S, Sturt NJ, Michils G, Tejpar S, Phillips RK, et al. Assessment of endostatin gene therapy for familial adenomatous polyposis-related desmoid tumors. Cancer Res. 2006;66(16):8233–40. https://doi.org/10.1158/0008-5472.Can-06-1209.
McGuinness LA, Higgins JPT. Risk-of-bias VISualization (robvis): an R package and Shiny web app for visualizing risk-of-bias assessments. Res Synth Meth. 2020. https://doi.org/10.1002/jrsm.1411.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Conflicts of interests/competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
TDM completed the literature search, wrote the first draft of the manuscript and critically revised the manuscript. SD and CDB contributed ideas and content and critically revised the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
McLean, T.D., Duchi, S. & Di Bella, C. Molecular Pathogenesis of Sporadic Desmoid Tumours and Its Implications for Novel Therapies: A Systematised Narrative Review. Targ Oncol 17, 223–252 (2022). https://doi.org/10.1007/s11523-022-00876-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-022-00876-z