
Abstract
Background
Preliminary data suggest that combining savolitinib, a potent and highly selective MET-tyrosine kinase inhibitor (TKI), with osimertinib, a third-generation, irreversible, oral epidermal growth factor receptor-TKI (EGFR-TKI), may overcome MET-based resistance to EGFR-TKIs.
Objective
To investigate the safety and tolerability of savolitinib in Japanese patients with advanced solid malignancies.
Patients and Methods
In Part C of the phase Ib, multi-arm, open-label, multicenter TATTON study, two cohorts of Japanese adult patients were evaluated across six study centers in Japan. Patients with advanced solid malignancies received oral savolitinib monotherapy 400 mg once daily (qd), escalating to 600 mg; patients with advanced EGFR mutation-positive (EGFRm) non-small-cell lung carcinoma (NSCLC) who progressed on prior EGFR-TKI received oral osimertinib 80 mg+savolitinib 300/400/600 mg qd combination therapy. Primary endpoints: safety/tolerability of savolitinib±osimertinib, and maximum tolerated dose(s) (MTD) definition.
Results
Seventeen patients received monotherapy; 12 received combination. Dose-limiting toxicities (DLTs): with monotherapy, 400 mg, none reported; 600 mg, n = 3/9 evaluable patients (33%) reported DLTs (grade 3 and 4 alanine aminotransferase and aspartate transaminase increased, and grade 4 drug-induced liver injury). With combination: 400 mg, 1/6 (17%) reported DLTs (grade 2 fatigue, nausea, and myalgia); 300 mg, none reported; 600 mg, 3/4 (75%) reported DLTs (grade 2 pyrexia, grade 3 skin reaction, and anaphylactic shock). Grade ≥3 adverse events were reported in 41% of patients receiving monotherapy and 33% receiving combination. TATTON is no longer recruiting patients.
Conclusions
The MTD of savolitinib was 400 mg qd in both cohorts. Data demonstrate an acceptable safety profile for savolitinib alone, or with osimertinib.
Trial registration: Clinicaltrials.gov; NCT02143466; 21 May 2014.
Plain Language Summary
For patients with epidermal growth factor receptor mutation-positive (EGFRm) non-small cell lung cancer (NSCLC), EGFR tyrosine kinase inhibitors, like osimertinib, are the standard treatment. However, for most patients, these treatments eventually stop working, as tumors develop resistance to them. Early studies suggest that combining osimertinib with savolitinib can overcome this resistance. We report Part C of the four-part TATTON study, in which two groups of Japanese adult patients received treatment. One group received savolitinib 400 mg once daily, then 600 mg. The other group received osimertinib 80 mg with savolitinib 300/400/600 mg once daily. The main objective of the study was to determine the maximum dose of savolitinib that patients could receive (maximum tolerated dose) and to monitor the safety of the combination. Overall, 17 patients received savolitinib alone and 12 received the combination. The maximum tolerated dose of savolitinib was found to be 400 mg once daily in both groups of patients. The data demonstrated that savolitinib had acceptable safety outcomes either alone, or in combination with osimertinib.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.14139947. |
In this phase Ib, multi-arm, open-label, multicenter study, the maximum tolerated dose of savolitinib was found to be 400 mg per day in the two study patient groups: patients with advanced solid malignancies receiving savolitinib monotherapy and patients with EGFRm NSCLC receiving osimertinib/savolitinib combination therapy. |
The study demonstrated an acceptable safety profile for savolitinib either alone or in combination with osimertinib. |
1 Introduction
Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) are the standard first-line therapy for patients with advanced EGFR-mutation positive (EGFRm) non-small-cell lung cancer (NSCLC) [1,2,3]. However, most patients with EGFRm NSCLC will eventually develop resistance to EGFR-TKIs through various mechanisms, including acquired EGFR resistance mutations, such as EGFR T790M, which is observed in approximately 50% of patients who progress on first- or second-generation EGFR-TKIs [4]. Resistance can also emerge via off-target genetic alterations such as MET amplification, which along with MET-based resistance mechanisms can occur in > 5% of patients progressing on first- or second-generation EGFR-TKIs [5,6,7].
Osimertinib is a third-generation, irreversible, oral EGFR-TKI that potently and selectively inhibits both EGFR mutation and EGFR T790M resistance mutations, and has demonstrated efficacy in NSCLC central nervous system metastases [8,9,10,11,12,13]. Osimertinib is currently approved in more than 85 countries, including the USA, Japan, and countries of the European Union, for the treatment of patients with advanced EGFR T790M mutation-positive NSCLC [14,15,16]. It is the recommended first-line treatment for patients with advanced EGFRm NSCLC, or for patients with an EGFR T790M mutation following disease progression on first- or second-generation EGFR-TKI therapy [1, 2]. In addition, adjuvant osimertinib has demonstrated a statistically significant and clinically meaningful improvement in disease-free survival in patients with non-squamous stage IB / II / IIIA EGFRm NSCLC [17].
In those patients who acquire MET-based resistance to EGFR-TKIs, preclinical and some early clinical data suggest that combined treatment with an EGFR-TKI and a MET inhibitor is an effective treatment option [7, 18, 19]. Savolitinib is an oral, potent, and highly selective MET-TKI demonstrating preliminary clinical activity in advanced solid tumors [20,21,22,23]. Therefore, combination of osimertinib and savolitinib could potentially improve outcomes and/or delay disease progression in patients with advanced EGFRm NSCLC.
The combination of osimertinib and savolitinib has been evaluated in the global phase Ib TATTON study (NCT02143466) [24, 25]. In the previously published TATTON Part A (dose-escalation cohort) [24], a dosing strategy with acceptable tolerability was identified for osimertinib plus savolitinib in non-Japanese patients with advanced EGFRm NSCLC and disease progression on a prior EGFR-TKI [24]. The efficacy and safety of this combination was further evaluated in the dose-expansion cohorts of TATTON Parts B/D, which has also previously been published [25]. Parts B/D enrolled patients with advanced, MET-amplified, EGFRm NSCLC. The results from these dose-expansion cohorts showed acceptable safety and promising anti-tumor activity in patients with EGFRm, MET-amplified NSCLC, who had disease progression on previous EGFR-TKI therapy [25].
Here, we report the results from Part C of the TATTON study, in which all of the enrolled patients were Japanese. Previously, studies have shown that Japanese patients with advanced EGFRm NSCLC benefit from treatment with osimertinib when received in either the first or second line [26, 27]. We present data on the safety, preliminary efficacy, and pharmacokinetic (PK) outcomes of savolitinib monotherapy in Japanese patients with advanced solid tumors and of osimertinib plus savolitinib in Japanese patients with advanced EGFRm NSCLC, whose disease had progressed on previous EGFR-TKI treatment.
2 Materials and Methods
2.1 Trial Design and Treatments
TATTON is a phase Ib, multi-arm, open-label, multicenter study divided into four Parts, A to D, to assess the safety, tolerability, and anti-tumor activity of osimertinib in combination with ascending doses of investigational agents in patients with EGFRm advanced NSCLC whose disease has progressed after EGFR-TKI therapy [24, 25]. Part C of the study, reported here, consisted of dose-finding and dose expansion cohorts, across six study centers in Japan: savolitinib monotherapy in patients with advanced solid tumors, and combination therapy with savolitinib and osimertinib in patients with EGFRm advanced NSCLC whose disease had progressed on a prior EGFR-TKI.
In the monotherapy cohorts, patients with advanced solid malignancies received oral savolitinib 400 mg once daily (qd) (AstraZeneca, Södertälje, Sweden), escalating to 600 mg qd. In the combination therapy cohorts, patients with advanced EGFRm NSCLC received oral osimertinib 80 mg qd (AstraZeneca, Södertälje, Sweden) plus savolitinib 400 mg qd. However, further to emerging results, the protocol was subsequently amended to include weight-based combination dosing where osimertinib 80 mg qd was combined with savolitinib 600 mg qd in patients with a body weight > 55 kg at enrollment, and savolitinib 300 mg qd in patients with a body weight ≤ 55 kg at enrollment. Hence more than six patients were enrolled at a nominal 600 mg dose level in some cohorts in Part C. Each patient received continuous administration of monotherapy or combination therapy qd for 28-day cycles.
Patients continued to receive treatment following disease progression if they were deemed, by the investigator, to be receiving clinical benefit in the absence of discontinuation criteria. Patients could have treatment discontinued for a variety of reasons, including adverse events (AEs), severe non-compliance with the protocol as judged by the investigator, or study sponsor and disease progression as per RECIST 1.1. If a patient receiving combination therapy discontinued osimertinib due to toxicity, they could continue to receive savolitinib monotherapy until disease progression. If a patient receiving combination therapy discontinued savolitinib due to toxicity, they could continue to receive osimertinib as monotherapy until the investigator deemed there to be no further benefit.
Patients provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki Good Clinical Practice guidelines (as defined by the International Conference on Harmonisation), applicable regulatory requirements, the Good Clinical Practice for Trials on Drugs’ (Japanese Ministry of Health, Labour, and Welfare Ordinance No. 28, 27 March 1997), and the policy on bioethics and human biological samples of the trial sponsor, AstraZeneca. Data underlying the findings described in this article may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
2.2 Patients
The study included patients from Japan aged 20 years or over, with World Health Organization performance status 0–1. Patients in the monotherapy group had advanced solid malignancies that were refractory to standard therapies or for which no standard therapies existed, excluding lymphoma, with radiological disease progression on the last treatment administered prior to enrolling in the study. Patients were also required to have evaluable or measurable disease as per RECIST. Patients in the combination group had histological or cytological confirmation of advanced NSCLC, with EGFR mutation known to be associated with EGFR-TKI sensitivity (including exon 19 deletion and L858R; EGFRm), and radiological disease progression on a prior EGFR-TKI treatment. Patients were not selected for MET. Patients had to have at least one lesion, not previously irradiated or biopsied during the screening period, that could be accurately measured at baseline as ≥ 10 mm in the longest diameter (except lymph nodes, which must have a short axis ≥ 15 mm) with computerized tomography or magnetic resonance imaging.
Exclusion criteria included patients with: any cytotoxic chemotherapy or anti-cancer drugs ≤ 14 days (or radiotherapy ≤ 1 week) before starting study treatment; any unresolved toxicity of grade > 1 from prior therapy (with the exception of alopecia and grade 2, prior platinum-therapy related neuropathy); any prior treatment with savolitinib; spinal cord compression or brain metastases unless asymptomatic, stable, and not requiring steroids for ≥ 2 weeks prior to study treatment; evidence of severe or uncontrolled systemic disease and/or active infection; inadequate liver or renal function; and/or cardiac abnormalities. In the combination group, treatment with an EGFR-TKI within approximately 5 × half-life of first study dose (≤ 8 days for erlotinib, gefitinib, or afatinib, or ≤ 10 days for dacomitinib) was not permitted; prior or current treatment with osimertinib or MET inhibitors (e.g. foretinib, crizotinib, cabozantinib, onartuzumab) was not allowed.
2.3 Objectives and Assessments
The primary objectives were safety and tolerability of savolitinib with or without osimertinib, and to define the combination dose(s) for further clinical evaluation.
A secondary objective in the monotherapy and combination therapy groups was to characterize the PK of savolitinib and/or osimertinib, and their metabolites, after single dosing and at steady state after multiple dosing. Secondary objectives in the combination group included preliminary assessment of anti-tumor activity by evaluation of tumor response, including objective response rate (ORR), duration of response (DoR), change in tumor size, and progression-free survival (PFS) per RECIST 1.1. ORR was defined as the percentage of patients who had at least one confirmed complete response (CR) or partial response (PR) prior to any evidence of progression (per RECIST 1.1). DoR was defined as the time from the date of first documented response until the date of documented progression or death in the absence of disease progression. PFS was defined as the time from the date of first dosing until the date of objective disease progression (per RECIST 1.1) or death (by any cause in the absence of progression), regardless of whether the patient withdrew from savolitinib therapy or received another anti-cancer therapy prior to progression.
AEs were collected from informed consent to the end of the follow-up period, defined as 28 days (±7 days) after study treatment was discontinued. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. For the monotherapy cohort, physical examinations were performed at screening, day 1 of cycles 1–8, and every 8 weeks until treatment discontinuation; clinical chemistry, liver function tests, and electrocardiograms were assessed at screening, cycle 1 (days 1, 8, 15, and 22), day 1 of cycles 2–7, and every 8 weeks until treatment discontinuation. For the combination therapy cohort, all of the above parameters were recorded at screening, cycle 1 (days 1, 8, 15, and 22), day 1 of cycles 2–7, and every 8 weeks until discontinuation. A Safety Review Committee evaluated the safety, tolerability, and PK (if available) of each cohort of savolitinib and confirmed the tolerated dose and the dose for further clinical evaluation in Japanese patients.
Tumor assessments were performed using computerized tomography or magnetic resonance imaging. Baseline assessments were performed no more than 28 days before the start of study treatment, with follow-up assessments every 6 weeks (±7 days) after the start of study treatment until cycle 7, and then every 8 weeks until disease progression as defined by RECIST 1.1, even if the patient discontinued treatment (unless the patient withdrew their consent).
Blood samples were collected pre-dose and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h after dosing for assessment of savolitinib and osimertinib single-dose (cycle 1 day 1) and multiple-dose PK (cycle 1 day 15 for monotherapy cohorts and cycle 2 day 1 for combination cohorts).
2.4 Statistical Analysis
Patients with advanced solid malignancies were enrolled to ensure a minimum of three and a maximum of six evaluable patients in each cohort. The total sample size varied, depending on the number of dose levels evaluated and the number of dose limiting toxicities (DLTs) observed. DLTs were defined as an AE or abnormal laboratory value that occurred from the first dose of study treatment up to the last day of cycle 1, assessed as unrelated to disease progression, intercurrent illness, or concomitant medications that despite optimal therapeutic intervention met any of the pre-defined criteria (Online Supplementary Material (OSM) – Methods). It was anticipated that there would be one or two dose levels in both cohorts; thus, approximately six to 12 evaluable patients were required for the monotherapy cohort, and approximately 12 evaluable patients were required for the combination therapy cohort. The number of patients was chosen to obtain adequate tolerability, safety, and PK data while exposing as few patients as possible to the investigational product and procedures.
Safety and efficacy were assessed in the safety analysis set, i.e. all patients who received at least one dose of study treatment. PK was assessed in the PK analysis set, i.e. all patients who received at least one dose of either combination treatment as per the study protocol, for whom any post-dose PK data were available and did not violate or deviate from the study protocol in ways that could significantly affect the PK analyses. The Kaplan-Meier method was used to analyze DoR and PFS. The individual savolitinib and osimertinib PK parameters were calculated by Covance Incorporated using Phoenix WinNonlin (version 8.1). Statistical analyses and summary statistics of PK parameters were performed in SAS (version 9.1.3).
3 Results
3.1 Patient Disposition and Baseline Characteristics
Between 19 February 2015 and 22 August 2019, 17 patients received savolitinib monotherapy (400 mg, n = 7; 600 mg, n = 10) and 12 patients received combination therapy with osimertinib and savolitinib (savolitinib dose 400 mg, n = 6; weight-based dosing: 300 mg, n = 2; 600 mg, n = 4) (Fig. 1).

Patient disposition. “Other” included progression of disease clinically. AE adverse event, PD progressive disease, PK pharmacokinetics
In the monotherapy cohorts, all 17 patients discontinued savolitinib, 13 (76%) due to progressive disease and four (24%) due to an AE (Fig. 1). The majority of patients had lung adenocarcinoma histology (n = 12, 71%) and metastatic disease (n = 12, 71%) (Table 1). Nine patients (53%) received more than three regimens of previous anti-cancer treatment; 11 (65%) patients previously received first- and second-generation EGFR-TKIs, while all patients (n = 17, 100%) also received cytotoxic chemotherapy, including 15 (88%) patients who received platinum-based doublet chemotherapy (Table 1).
In the combination therapy cohorts, three out of 12 (25%) patients were still receiving both treatments at the time of data cut-off (Fig. 1). Nine of the 12 (75%) patients discontinued savolitinib, six (50%) due to an AE and three (25%) due to progressive disease; eight (67%) patients discontinued osimertinib, six (50%) due to progressive disease, one (8%) due to an AE, and one (8%) due to progression clinically reported as “other” (Fig. 1). Like the monotherapy cohort, the majority of patients had lung adenocarcinoma histology (n = 12, 100%) and metastatic disease (n = 8, 67%) (Table 1). In the combination therapy cohorts, half (n = 6, 50%) of patients received only one regimen of previous anti-cancer treatment; all patients (n = 12, 100%) received first- and second-generation EGFR-TKIs as per eligibility criteria and seven (58%) patients received cytotoxic chemotherapy (Table 1).
3.2 Safety
The incidence and type of DLTs were recorded in each cohort for each dose of savolitinib (Fig. 2). In the monotherapy cohort, for patients receiving savolitinib 400 mg, no DLTs were reported but 3/9 evaluable patients (33%) receiving savolitinib 600 mg reported DLTs (one patient had grade 3 alanine aminotransferase (ALT) and aspartate aminotransferase (AST) increased, one patient had grade 4 ALT and AST increased, and one patient had grade 4 drug-induced liver injury). In the combination cohort, for patients receiving savolitinib 400 mg, 1/6 (17%) reported DLTs (fatigue, nausea, and myalgia (all grade 2)). For the weight-based dosing cohort, 3/4 (75%) of the evaluable patients reported DLTs, all of whom were receiving the savolitinib 600 mg dose (grade 2 pyrexia, grade 3 skin reaction, and grade 3 anaphylactic shock, in one patient each).

Dose-limiting toxicities (DLTs) across doses in (a) monotherapy in patients with advanced solid malignancies and (b) combination therapy in patients with epidermal growth factor receptor mutation positive advanced non-small cell lung cancer. ALT alanine aminotransferase, AST aspartate aminotransferase. *Not recovered/not resolved
The median actual treatment durations of savolitinib in the monotherapy and combination cohorts were 1.4 months (range: 0.2–15.7 months) and 3.0 months (range: 0.4–20.3 months), respectively, and for osimertinib in the combination cohort was 6.0 months (range: 0.8–20.3 months).
In the monotherapy cohorts, one (6%) patient experienced one dose reduction due to an AE during the first 28 days of treatment. During the rest of the study, one (6%) patient experienced a dose interruption and two (12%) patients experienced dose reductions, due to an AE in all cases. Dose re-escalations were not permitted in the protocol.
In the combination therapy cohorts for savolitinib dosing, one (8%) patient experienced one dose reduction due to their own decision and three (25%) patients experienced one dose interruption, each due to AEs during the first 28 days of treatment. During the rest of the study, two (17%) patients experienced one dose interruption each, and three (25%) patients experienced one dose reduction each due to AEs in all cases (one patient experienced both a dose reduction and a dose interruption). For osimertinib dosing, during the first 28 days of treatment, no patients experienced dose reductions and five (42%) patients experienced one (n = 4, 33%) or two (n = 1, 8%) dose interruptions due to AEs. During the rest of the study, three (25%) patients experienced one dose interruption each due to AEs.
In the monotherapy cohorts, 16 (94%) patients reported at least one AE during the study (Table 2). The most common AEs, regardless of grade or cause, were nausea (n = 10, 59%) and vomiting (n = 7, 41%) (Table 3). Grade ≥ 3 AEs were reported in seven (41%) patients, comprising ALT increase (n = 3, 18%), AST increase (n = 3, 18%), drug hypersensitivity (n = 1, 6%), drug-induced liver injury (n = 1, 6%), lung infection (n = 1, 6%), maculopapular rash (n = 1, 6%), neutrophil count decrease (n = 1, 6%), pyrexia (n = 1, 6%), and white blood cell count increase (n = 1, 6%) (Table 3). Serious AEs (SAEs) were reported in five (29%) patients (Table 4). Four (24%) patients experienced AEs leading to discontinuation of savolitinib, of which three (18%) of these patients had AEs that were considered possibly causally related to savolitinib and comprised drug hypersensitivity, drug-induced liver injury, and ALT and AST increase (AEs occurred once in one patient each). No patient in the monotherapy cohort died during the study as a result of an AE while one patient in the monotherapy cohort died of disease progression during the follow-up period (36 days after the dose of study treatment), considered possibly causally related to the disease under investigation.
In the combination cohorts, all patients reported at least one AE during the study (Table 2). The most common AEs regardless of grade or cause were diarrhea (n = 6, 50%) and pyrexia (n = 5, 42%) (Table 3). Four (33%) patients reported grade ≥ 3 AEs, which were anaphylactic shock, dental caries, bile duct stone, and skin reaction (all occurring once in one patient each) (Table 3). SAEs were reported in three (25%) patients (Table 4). Six (50%) patients experienced AEs leading to discontinuation of savolitinib in the combination therapy cohorts, all of which were considered possibly causally related to savolitinib, and were anaphylactic shock (n = 1, 8%), headache (n = 1, 8%), skin reaction (n = 1, 8%), myalgia (n = 1, 8%), peripheral edema (n = 1, 8%), and pyrexia (n = 2, 17%); the event of myalgia also led to discontinuation of osimertinib. No patient in the combination cohort died during the study as a result of an AE.
3.3 Efficacy
None of the 17 patients with advanced solid tumors across the monotherapy cohorts had an objective response. Nine (53%) patients had stable disease (SD) ≥ 6 weeks and eight (47%) patients had disease progression (Table 5). For the patients with EGFRm NSCLC receiving combination therapy, ORR was 42% (95% confidence interval [CI] 15.2–72.3), with five of the 12 patients having a PR. Of the seven non-responders, six patients had SD ≥ 6 weeks and one patient was “not evaluable” (Table 5).
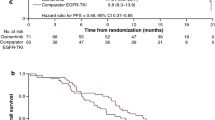
In the combination therapy cohorts, the median DoR was 3.1 months (95% CI 2.7–not reached); of the five patients with a PR, 3 (60%) were still in response after 3 months and two (40%) were still in response after 12 months; the maturity of the response data was 60% (Table 5). Median PFS (across doses) in the monotherapy (94% maturity) and combination therapy (50% maturity) cohorts were 2.6 months (95% CI 1.4–3.2) and 5.5 months (95% CI 3.9–not reached), respectively (Table 5). Best percent change from baseline in target lesion size for both cohorts is shown in Fig. 3.

Waterfall plot of the best percentage change in target lesion size in (a) patients with advanced solid malignancies receiving savolitinib monotherapy and (b) patients with epidermal growth factor receptor mutation positive advanced non-small-cell lung cancer receiving savolitinib + osimertinib combination therapy (by dose of savolitinib)
3.4 Pharmacokinetics
Savolitinib was rapidly absorbed and eliminated with a mean half-life of 3.30–4.93 h with no accumulation following multiple dosing and similar PK parameters following monotherapy and combination with osimertinib (OSM – Results, Table S1). Following osimertinib 80 mg qd dosing, osimertinib PK parameters were consistent regardless of the savolitinib combination dose, with 1.26- to 7.59-fold accumulation at steady state (OSM, Table S1).
4 Discussion
MET amplification is a common mechanism of acquired resistance in EGFRm tumors treated with EGFR-TKIs [6, 28,29,30,31]. The purpose of Part C of the TATTON study was to investigate the safety and tolerability of savolitinib as a monotherapy and in combination with osimertinib in Japanese patients. These preliminary data demonstrate an acceptable safety profile for savolitinib alone and in combination with osimertinib in Japanese patients with advanced EGFRm NSCLC.
As a third of patients in the monotherapy cohort receiving 600 mg savolitinib as a single daily dose reported a DLT, compared with none reported at 400 mg, it was determined that 600 mg qd was not tolerable in this Japanese patient population. Similarly, for the combination study, 600 mg qd was received due to weight-based dosing and was deemed not tolerable, as 75% of evaluable patients reported DLTs. Thus, in this study, savolitinib at once-daily doses up to 400 mg, administered as monotherapy in Japanese patients with advanced solid tumors and in combination with osimertinib 80 mg in Japanese patients with EGFRm NSCLC, demonstrated an acceptable safety profile. This was therefore determined to be the maximum tolerated dose (MTD) for combination treatment. Overall, safety outcomes were in line with data from the global cohorts in TATTON Parts A, B, and D, wherein the most frequent AEs in this study, such as diarrhea and nausea, were also the most common AEs in the savolitinib/osimertinib arms in those studies. Also, as in the prior TATTON parts, tolerability of the combination therapy was improved with the lower savolitinib dose [24, 25]. In addition, the safety profile of the monotherapy cohort aligns with that seen in the phase I study, in which patients with advanced solid tumors received savolitinib [22].
With regard to the anti-cancer efficacy of savolitinib, no responses were observed in the monotherapy cohort, as might be expected from historical data for a targeted agent in an unselected advanced solid tumor treatment-refractory phase I patient population [22]. For combination therapy, our preliminary data suggest anti-tumor activity in this more selected Japanese population with advanced EGFRm NSCLC, in line with data from the prior TATTON cohorts, Parts A, B, and D, in which efficacy was comparable when osimertinib was combined with 300 mg qd or 600 mg qd of savolitinib [24, 25]. However, it should be noted that, per protocol, the patients were unselected for MET or T790M status. In addition, the PK data showed that exposure of savolitinib in combination was broadly similar to monotherapy data with no apparent PK interaction in Japanese patients. This is consistent with the conclusions from TATTON Part A in non-Japanese patients, where the PK parameters of savolitinib and osimertinib in combination were within the ranges observed for their respective monotherapy treatment [24]. In general, no substantial differences in the steady-state PK parameters for savolitinib and osimertinib were observed in Japanese patients from TATTON Part C compared with non-Japanese patients in TATTON Part A [24]. Planchard et al. have similarly found no relevant differences in osimertinib steady-state AUC between Japanese and non-Japanese patients with NSCLC in a previous phase 1b study [32].
This study has several limitations that should be considered. Firstly, only 29 patients were assessed, making it difficult to extrapolate the data to a broader population. Furthermore, as per protocol, these patients had no MET-status data, so activity towards MET-amplified/overexpressed NSCLC cannot be concluded. Finally, patients who had no EGFR T790M status data at disease progression following EGFR-TKI therapy did not previously receive osimertinib in the EGFRm NSCLC cohort. This potentially makes the interpretation of the efficacy data difficult, as it cannot be conclusively stated that the treatment effect comes from the combination of osimertinib and savolitinib, and not just osimertinib alone.
In conclusion, these preliminary data demonstrate an acceptable safety profile for savolitinib (up to 400 mg as a single daily dose)/osimertinib combination therapy, indicating that it may be a potential treatment option for Japanese patients with EGFRm NSCLC whose disease had progressed on a prior EGFR-TKI. The combination savolitinib 300 mg plus osimertinib 80 mg is being investigated in the global phase II SAVANNAH trial (NCT03778229) in patients with EGFRm, MET-amplified/overexpressed advanced NSCLC following prior osimertinib [25, 33, 34], as well as in the ORCHARD (NCT03944772) phase II biomarker-directed, platform study [35].
References
Hanna N, Johnson D, Temin S, Baker S Jr, Brahmer J, Ellis PM, et al. Systemic therapy for stage IV non-small-cell lung cancer: American Society of Clinical Oncology Clinical Practice Guideline update. J Clin Oncol. 2017;35(30):3484–515. https://doi.org/10.1200/jco.2017.74.6065.
Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(suppl 4):iv192–iv237. doi: https://doi.org/10.1093/annonc/mdy275.
Wu YL, Planchard D, Lu S, Sun H, Yamamoto N, Kim DW, et al. Pan-Asian adapted Clinical Practice Guidelines for the management of patients with metastatic non-small-cell lung cancer: a CSCO-ESMO initiative endorsed by JSMO, KSMO, MOS, SSO and TOS. Ann Oncol. 2019;30(2):171–210. https://doi.org/10.1093/annonc/mdy554.
Wang ZF, Ren SX, Li W, Gao GH. Frequency of the acquired resistant mutation T790M in non-small cell lung cancer patients with active exon 19Del and exon 21 L858R: a systematic review and meta-analysis. BMC Cancer. 2018;18(1):148. https://doi.org/10.1186/s12885-018-4075-5.
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: https://doi.org/10.1126/scitranslmed.3002003.
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–7. https://doi.org/10.1158/1078-0432.CCR-12-2246.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43. https://doi.org/10.1126/science.1141478.
Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–61. https://doi.org/10.1158/2159-8290.CD-14-0337.
Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med. 2017;376(7):629–40. https://doi.org/10.1056/NEJMoa1612674.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–25. https://doi.org/10.1056/NEJMoa1713137.
Wu YL, Ahn MJ, Garassino MC, Han JY, Katakami N, Kim HR, et al. CNS efficacy of osimertinib in patients with T790M-positive advanced non-small-cell lung cancer: data from a randomized phase III trial (AURA3). J Clin Oncol. 2018;36(26):2702–9. https://doi.org/10.1200/jco.2018.77.9363.
Reungwetwattana T, Nakagawa K, Cho BC, Cobo M, Cho EK, Bertolini A, et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non–small-cell lung cancer. J Clin Oncol. 2018;36(33):3290–7. https://doi.org/10.1200/jco.2018.78.3118.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. https://doi.org/10.1056/NEJMoa1913662.
Japanese Ministry of Health LaW: TAGRISSO™ (osimertinib) Prescribing Information. http://med.astrazeneca.co.jp/product/brand-tag.html# (2018). Accessed 14 August 2020.
US Food & Drug Administration: TAGRISSO™ (osimertinib) Highlights of Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208065s007lbl.pdf (2018). Accessed 14 August 2020.
European Medicines Agency: TAGRISSO™ (osimertinib) Summary of Product Characteristics. https://www.ema.europa.eu/documents/product-information/tagrisso-epar-product-information_en.pdf (2018). Accessed 14 August 2020.
Wu Y-L, Tsuboi M, He J, John T, Grohe C, Majem M, et al. Osimertinib in resected EGFR-mutated non–small-cell lung cancer. NEJM. 2020;383(18):1711–23. https://doi.org/10.1056/NEJMoa2027071.
Gainor JF, Niederst MJ, Lennerz JK, Dagogo-Jack I, Stevens S, Shaw AT, et al. Dramatic response to combination erlotinib and crizotinib in a patient with advanced, EGFR-mutant lung cancer harboring de novo MET amplification. J Thorac Oncol. 2016;11(7):e83–5. https://doi.org/10.1016/j.jtho.2016.02.021.
York ER, Varella-Garcia M, Bang TJ, Aisner DL, Camidge DR. Tolerable and effective combination of full-dose crizotinib and osimertinib targeting MET amplification sequentially emerging after T790M positivity in EGFR-mutant non-small cell lung cancer. J Thorac Oncol. 2017;12(7):e85–8. https://doi.org/10.1016/j.jtho.2017.02.020.
Jia H, Dai G, Weng J, Zhang Z, Wang Q, Zhou F, et al. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2, 3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J Med Chem. 2014;57(18):7577–89. https://doi.org/10.1021/jm500510f.
Gavine PR, Ren Y, Han L, Lv J, Fan S, Zhang W, et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9(1):323–33. https://doi.org/10.1016/j.molonc.2014.08.015.
Gan HK, Millward M, Hua Y, Qi C, Sai Y, Su W, et al. First-in-human phase I study of the selective MET inhibitor, savolitinib, in patients with advanced solid tumors: safety, pharmacokinetics, and antitumor activity. Clin Cancer Res. 2019;25(16):4924–32. https://doi.org/10.1158/1078-0432.Ccr-18-1189.
Hua Y, Shen L, Gan H, Lickliter J, Millward M, Xu J, et al. Abstract CT305: phase I studies of a selective cMet inhibitor AZD6094 (HMPL504/volitinib) in patients with advanced solid tumors. Cancer Res. 2015;75(suppl 15):CT305. doi: https://doi.org/10.1158/1538-7445.AM2015-CT305.
Oxnard GR, Yang JC, Yu H, Kim SW, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31(4):507–16. https://doi.org/10.1016/j.annonc.2020.01.013.
Sequist LV, Han J-Y, Ahn M-J, Cho BC, Yu H, Kim S-W, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020;21(3):373–86.
Ohe Y, Imamura F, Nogami N, Okamoto I, Kurata T, Kato T, et al. Osimertinib versus standard-of-care EGFR-TKI as first-line treatment for EGFRm advanced NSCLC: FLAURA Japanese subset. Jpn J Clin Oncol. 2019;49(1):29–36. https://doi.org/10.1093/jjco/hyy179.
Akamatsu H, Katakami N, Okamoto I, Kato T, Kim YH, Imamura F, et al. Osimertinib in Japanese patients with EGFR T790M mutation-positive advanced non-small-cell lung cancer: AURA3 trial. Cancer Sci. 2018;109(6):1930–8. https://doi.org/10.1111/cas.13623.
Papadimitrakopoulou VA, Wu YL, Han JY, Ahn MJ, Ramalingam SS, John T, et al. LBA51 Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29(suppl 8):mdy424.064. https://doi.org/10.1093/annonc/mdy424.064.
Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4(11):1527–34. https://doi.org/10.1001/jamaoncol.2018.2969.
Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8(12):1529–39. https://doi.org/10.1158/2159-8290.Cd-18-1022.
Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29(suppl 8):viii740.
Planchard D, Brown KH, Kim DW, Kim SW, Ohe Y, Felip E, et al. Osimertinib Western and Asian clinical pharmacokinetics in patients and healthy volunteers: implications for formulation, dose, and dosing frequency in pivotal clinical studies. Cancer Chemotherap Pharmacol. 2016;77(4):767–76. https://doi.org/10.1007/s00280-016-2992-z.
Oxnard GR, Cantarini M, Frewer P, Hawkins G, Peters J, Howarth P, et al. SAVANNAH: A phase II trial of osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-driven (MET+), locally advanced or metastatic non-small cell lung cancer (NSCLC), following disease progression on osimertinib. J Clin Oncol. 2019;37(suppl 15):TPS9119. https://doi.org/10.1200/JCO.2019.37.15_suppl.TPS9119.
Ahn M, Cantarini M, Frewer P, Hawkins G, Peters J, Howarth P, et al. P1.01-134 SAVANNAH: phase II trial of osimertinib + savolitinib in EGFR-mutant, MET-driven advanced NSCLC, following prior osimertinib. J Thorac Oncol. 2019;14(10):S415–S6. doi: https://doi.org/10.1016/j.jtho.2019.08.849.
Yu H, Goldberg SB, Le X, Piotowska Z, Smith P, Mensi I, et al. ORCHARD: a phase II platform study in patients with advanced EGFRm NSCLC who have progressed on first-line osimertinib therapy. Poster P201-22 presented at the IASLC World Conference of Lung Cancer (WCLC), Barcelona, Spain, 7–10 September 2019.
Acknowledgements
The authors thank all the patients and their families. They would also like to acknowledge Bernadette Tynan, MSc, of Ashfield MedComms, an Ashfield Health company, for medical writing support that was funded by AstraZeneca (Cambridge, UK) in accordance with Good Publications Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study (NCT02143466) was funded by AstraZeneca (Cambridge, UK), the manufacturers of savolitinib and osimertinib.
Conflicts of interest
KY received honoraria and research funds from AstraZeneca. TH received honoraria from Ono, Eli Lilly, AstraZeneca, Taiho, Chugai, MSD and Boehringer Ingelheim; research funds from Ono, Eli Lilly, AstraZeneca, Taiho, Chugai, Merck Serono, MSD and Boehringer Ingelheim. HS received honoraria from AstraZeneca and research funds from MSD, AstraZeneca, BMS, Takeda, Ono and Parexel. TK received honoraria from AstraZeneca, Eli Lilly, MSD, Ono, Boehringer Ingelheim, Chugai and Bristol Myers; research funds from Bristol Myers, Takeda, MSD, AstraZeneca and Novartis. YO received honoraria from AstraZeneca and Chugai; research funds from AstraZeneca, Chugai, ONO, BMS, Kyorin, Dainippon- Sumitomo, Taiho, Kissei, Ignyta, Takeda, Janssen and LOXO. TH received honoraria and research funds from AstraZeneca and Novartis. AM reports employment for AstraZeneca. RBV reports former employment for AstraZeneca and current employment for Johnson & Johnson; holding shares of AstraZeneca, Aduro Biotech and Johnson & Johnson. GFA reports former employment for AstraZeneca and current employment for UCB Pharma Ltd. MH, XO, KS report employment for AstraZeneca. GO reports employment for Foundation Medicine and honoraria from AstraZeneca.
Ethics approval
The study was conducted in accordance with the Declaration of Helsinki Good Clinical Practice guidelines (as defined by the International Conference on Harmonisation), applicable regulatory requirements, the Good Clinical Practice for Trials on Drugs’ (Japanese Ministry of Health, Labour, and Welfare Ordinance No. 28, 27 March 1997), and the policy on bioethics and human biological samples of the trial sponsor, AstraZeneca.
Consent to participate
Patients provided written informed consent.
Availability of data and material
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Authors’ contributions
Conception and design: Ghada Ahmed, Anders Mellemgaard, Geoffrey Oxnard. Collection and assembly of data: Kiyotaka Yoh, Tomonori Hirashima, Hideo Saka, Takayasu Kurata, Yuichiro Ohe, Toyoaki Hida, Anders Mellemgaard, Remy Verheijen. Data analysis and interpretation: All authors. Drafting the manuscript: All authors. Manuscript writing: No authors, see medical writing acknowledgement. All authors read and approved the final draft of the manuscript. All authors agree to be accountable for all aspects of the work, which includes ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Yoh, K., Hirashima, T., Saka, H. et al. Savolitinib ± Osimertinib in Japanese Patients with Advanced Solid Malignancies or EGFRm NSCLC: Ph1b TATTON Part C. Targ Oncol 16, 339–355 (2021). https://doi.org/10.1007/s11523-021-00806-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-021-00806-5