
Abstract
There are strong biologic and preclinical rationales for the development of therapeutic cancer vaccines; however, the clinical translation of this treatment strategy has been challenging. It is now understood that many previous clinical trials of cancer vaccines used target antigens or vaccine designs that inherently lacked sufficient immunogenicity to induce clinical responses. Despite the historical track record, breakthrough advances in cancer immunobiology and vaccine technologies have supported continued interest in therapeutic cancer vaccinations, with the hope that next-generation vaccine strategies will enable patients with cancer to develop long-lasting anti-tumor immunity. There has been substantial progress identifying antigens and vaccine vectors that lead to strong and broad T cell responses, tailoring vaccine designs to achieve optimal antigen presentation, and finding combination partners employing complementary mechanisms of action (e.g., checkpoint inhibitors) to overcome the diverse methods cancer cells use to evade and suppress the immune system. Results from randomized, phase 3 studies testing therapeutic cancer vaccines based on these advances are eagerly awaited. Here, we summarize the successes and failures in the clinical development of cancer vaccines, address how this historical experience and advances in science and technology have shaped efforts to improve vaccines, and offer a clinical perspective on the future role of vaccine therapies for cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Clinical translation of vaccine therapies for cancer has been challenging due to the complexity of cancer immunology and optimal vaccine design. |
Advances in vaccine technology and understanding of cancer immunology support continued investigation of vaccine-based treatment strategies for cancer. |
1 Introduction
Successful cancer immunotherapy ultimately requires tumor cell engagement by cytolytic effectors (T cells and antibodies) capable of specifically recognizing unique or aberrantly expressed tumor-associated antigens (TAAs) or tumor-specific antigens (TSAs). While some patients with cancer spontaneously generate sufficient levels or function of antigen-specific T cells with the potential to generate impressive anti-tumor activity, the majority do not. One approach to ensure an adequate level and function of immune effectors is through therapeutic cancer vaccination. This form of active immunotherapy aims to generate anti-tumor immune responses directed against TAAs or TSAs [1, 2]. The idea of vaccination against cancer has a long history and was initially built on the observation that some tumors spontaneously regress in patients experiencing an acute infection [3]. More than a century ago, Dr. William Coley leveraged this observation to develop a rudimentary anti-cancer immune therapy consisting of heat-inactivated bacteria [3, 4]. How a non-specific innate immune response against bacterial products could translate into a specific anti-tumor immune response was explained subsequently by the discovery that antigen-presenting cells (APCs) (dendritic cells [DCs]) could acquire immunogenic tumor-derived peptides released during the innate immune response. These peptides could then be used to activate anti-tumor T cells with cognate receptors [5]. This led to the hypothesis that use of tumor-derived antigens, if delivered to the immune system in a sufficiently immunogenic context (a “vaccine”), would, due to the preferential targeting of cancer cells, enable relatively safe and yet effective treatments for cancer, capable of inducing long-lasting immunity [6].
Despite this encouraging foundation, and although cancer vaccines have been the subject of intense preclinical and clinical investigation in a variety of malignancies over the past 40 years, the successful clinical translation from bench to bedside has been slow, with only two therapeutic cancer vaccines (sipuleucel-T and talimogene laherparepvec [T-VEC]) having gained regulatory approval in the United States or European Union and numerous negative phase 3 studies leading to product discontinuations. However, the interest in therapeutic cancer vaccination remains high for several reasons. First, the clinical efficacy of checkpoint inhibitors and the identification of tumor-antigen-specific T cells in treated patients now provide evidence that patients are able to prime tumor-reactive T cells and that this likely occurs spontaneously in the minority of cancer patients responding to checkpoint blockade monotherapy. Second, the identification of checkpoint-expressing T cells and checkpoint ligand-expressing tumor cells after cancer vaccine therapies suggests that combination therapies incorporating vaccines and checkpoint inhibitors may be effective, as demonstrated in preclinical studies [7,8,9,10]. Third, negative studies have provided lessons for the field moving forward, which are being applied in current trials and will be also used in future investigations. The main lessons, as recently reviewed by Hollingsworth and Jansen (2019), include the need for antigens and vaccine designs that elicit greater immunogenicity (particularly through optimal presentation of tumor antigens by professional DCs [6]) as well as combination treatment strategies to overcome multiple mechanisms of tumor-mediated immunosuppression [11]. Fourth, a deeper understanding of major histocompatibility complex (MHC)-antigen binding has evolved to allow for better vaccine design and selection of appropriate antigens. Fifth, the ability to preferentially induce type 1 anti-tumor immunity versus the more common type 2 tumor supportive immunity has increased. Finally, although limited efficacy has been observed with the therapeutic cancer vaccine sipuleucel-T, its approval provided clinical validation of the therapeutic vaccination concept, which remains scientifically sound.
Given how recent advances may transform the track record of cancer vaccines, there is a need to summarize these developments and how they will affect the future role of vaccines. This review describes the successes and failures in the clinical development of cancer vaccines, addresses how this historical experience and advances in science and technology have shaped efforts to improve vaccines (e.g., through optimizing antigen presentation by professional APCs), and offers a clinical perspective on the future role of vaccine therapies for cancer.
2 Historical Overview of Cancer Vaccines
Several types of cancer vaccines have been developed that vary depending on the form of the delivered antigen used in the vaccine: proteins or synthetic peptides of cancer antigens, cell-based delivery of tumor antigen (e.g., modified tumor cells, DCs loaded with tumor antigens), and DNA/RNA coding for cancer antigens (e.g., plasmids, RNA, viral vectors) (Fig. 1).

[Figure reproduced from Maeng H et al. F1000Res. 2019 https://doi.org/10.12688/f1000research.18693.1. Licensed under CC BY 4.0.] CD cluster of differentiation, IFN interferon, IL interleukin, IL2Rα IL-2 receptor alpha, MHC major histocompatibility complex, TCR T cell receptor
Diverse therapeutic cancer vaccine platforms have a common mechanism of action [12].
2.1 Peptide- and Protein-Based Vaccines
Peptide-based vaccines are relatively easy to manufacture, but combination with potent immune adjuvants is often needed to boost immunogenicity, and the number of people who may benefit from a given peptide vaccine is restricted by human leukocyte antigen (HLA) haplotype [13]. Several phase 3 studies investigating early peptide-based vaccines have not demonstrated clinical benefit despite demonstrating some induction of immune responses against TAAs or TSAs (Table 1) [11]. Explanations for lack of clinical benefit may lie in the properties of the peptides and adjuvants used, and early peptide vaccines may have been inherently inadequate for promoting antigen presentation and generating potent and durable anti-tumor immunity [6, 60,61,62].
A limitation of many early peptide vaccines was the use of short peptides (< 15 amino acids), including the minimal-length epitopes required to target cytotoxic lymphocytes (CTLs) but not T helper cells [6]. Short peptide epitopes are loaded onto non-professional APCs, including T cells and B cells [6, 63]. Yet, non-professional APCs circulate to non-inflamed lymphoid organs and do not deliver costimulatory signals to optimally prime and activate CTLs, thereby promoting tolerization [63]. Furthermore, cross-presentation of short peptides by professional APCs (DCs) is not as efficient or long lasting as that for synthetic long peptides [64]. Unfortunately, vaccines based on whole proteins (including idiotype vaccines) have also been largely unsuccessful in the clinic (Table 1). This may be due to the fact that the processing and presentation of whole proteins by DCs is inferior when compared with that for shorter peptides [65].
Overall, these results have provided rationale for the development of improved peptides such as synthetic long peptides with optimized immunogenicity, alternative peptide-delivery platforms such as nanoparticles, and more potent vaccine adjuvants.
2.2 Cellular Vaccines
Commonly studied types of cell-based cancer vaccines include DCs loaded with tumor (neo)antigens, modified autologous cancer cells, and allogeneic tumor cell lines. Cell-based vaccines were among the initial types of therapeutic cancer vaccines tested. The first therapeutic cancer vaccine approved by the United States Food and Drug Administration was sipuleucel-T, a vaccine consisting of autologous peripheral blood mononuclear cells, including DCs, loaded with the prostatic acid phosphatase antigen fused with granulocyte-macrophage colony-stimulating factor (GM-CSF; an immune-cell activator). The approval of sipuleucel-T in 2010 for metastatic castration-resistant prostate cancer was based on results from the phase 3 IMPACT trial (NCT00065442) showing that treatment with sipuleucel-T significantly improved overall survival (OS) compared with placebo (median 25.8 vs. 21.7 months; hazard ratio [HR] 0.78; p = 0.03; Table 2) [66]. These results and subsequent approval provided an early clinical validation of the therapeutic cancer vaccine concept. Real-world analyses suggest that sipuleucel-T remains an effective treatment option in the current treatment landscape, which includes androgen-receptor signaling pathway inhibitors (ASPIs). A retrospective cohort analysis of men with metastatic castration-resistant prostate cancer (N = 6044; January 2013–December 2017) found that treatment with sipuleucel-T as first-line therapy or any-line therapy was associated with improved OS compared with treatment with ASPIs alone [68].
In contrast, cellular vaccines based on autologous tumor cells have not had the same success in several pivotal trials, as they either did not meet their primary endpoints or were discontinued early because of clinical futility (Table 1). One possible explanation for this lack of success is the presence of immunosuppressive factors in the irradiated tumor cells or tumor cell lysates used for these vaccines [69, 70].
2.3 Genetic Vaccines
Viruses or plasmids can act as vectors for DNA or RNA encoding TAAs [11,12,13]. Viruses represent a promising platform for vaccines, as virus DNA or RNA may activate DCs by triggering pattern recognition receptors [11, 71].
As monotherapy, virus vector vaccines have not yet demonstrated consistent clinical benefit as demonstrated with the experiences with PROSTVAC and PANVAC (Table 1). For example, although a virus vector vaccine (PROSTVAC-VF) demonstrated OS benefit (but not progression-free survival [PFS; primary endpoint] or response) in a randomized phase 2 study of patients with metastatic castration-resistant prostate cancer [72], this positive signal was not validated in a subsequent phase 3 study (Table 1) [11, 16]. The investigators on the phase 3 study speculated that either PROSTVAC-VF did not generate sufficient immune responses as a single agent (possibly due to the choice of antigen or disease setting) or immunity was hampered by an immunosuppressive microenvironment [16]. To address these considerations, clinical trials of PROSTVAC-VF in combination with checkpoint inhibitors [11] and/or other cancer vaccines are ongoing (NCT02933255, NCT04020094, NCT03532217, and NCT03315871). However, a recent randomized phase 2 study found that addition of a viral vaccine to checkpoint inhibition did not yield a survival benefit in soft tissue sarcoma [17]. There is very limited clinical investigation of the PANVAC vaccine for pancreatic tumors (NCT00669734).
Like viral platforms, plasmid vector-based vaccines have intrinsic adjuvant immunogenicity because the nucleic acid itself may be immunostimulatory, triggering an innate immune response [73]. Other advantages of this platform are enhanced stability, ease of manufacturing, induction of intracellular antigen expression, and if full-length genes are utilized, there is no HLA restriction. Despite these advantages, there has been limited late-stage investigation of DNA or RNA vaccines to date. The DNA vaccine Allovectin-7®, which contains DNA sequences for HLA-B7 and β2 microglobulin, did not improve objective response rate or OS compared with chemotherapy in patients with advanced melanoma in a phase 3 trial, leading to termination of the development program. There have since been numerous improvements in DNA and RNA delivery technologies (e.g., electroporation) and more modern nucleic acid vaccines have been tested in early-stage studies, as described in Sect. 3.
2.4 Other Types of Cancer Vaccines
In contrast to the therapeutic cancer vaccines described previously, some vaccines used in the treatment of cancer do not deliver defined tumor antigens to generate anti-tumor immunity, but nevertheless, generate an immune response. For example, intravesical immunotherapy (i.e., vaccination) with Mycobacterium bovis bacillus Calmette–Guérin (BCG) is approved for the treatment of certain types of bladder cancer. The mechanism of action of BCG immunotherapy has not been well understood, but recent data indicate that BCG improves the activation and exhaustion status of tumor-specific T cells [74].
Another cancer vaccine approach involves strategies to modify or inflame tumor cells by intratumoral administration of oncolytic viruses. In 2015, the oncolytic viral vaccine T-VEC, a herpes virus genetically modified to express GM-CSF [13], was licensed for the treatment of patients with unresectable melanoma. The approval of T-VEC was based on the phase 3 OPTiM trial (NCT00769704) demonstrating that a higher proportion of patients treated with T-VEC versus GM-CSF had a durable clinical response (≥ 6 months continuously and beginning within the first year; 16.3% vs. 2.1%, respectively; p < 0.001; Table 2). There was also a trend for improved OS in the T-VEC group (23.3 vs. 18.9 months; p = 0.051) [67]. The mechanism of action for oncolytic viral vaccines such as T-VEC is different to that of other notable cancer vaccines. This form of “in situ” vaccination results in the killing of tumor cells by the virus and the release of tumor antigens [75, 76]. These effects lead to the immune-mediated regression of distant tumor lesions, presumably either through amplification of previously activated host-immunity and/or the priming of new anti-tumor immune responses [75,76,77]. Evidence for tumor-specific T cell induction after T-VEC treatment was observed in a clinical study of patients with stage IIIc and stage IV melanoma. In this study, a patient with a complete response after T-VEC injection had an increase in MART-1-specific effector T cells, both in the injected target lesion and in a nontarget lesion [77].
3 The Evolution of Cancer Vaccine Development: Current Strategies Based on Historical Experience and Scientific Advances
The limited success of therapeutic cancer vaccines despite decades of development by academia and industry raises the questions of why expectations have not been fulfilled and how barriers to successful development can be overcome. Advances in our understanding of antigen immunogenicity, the importance of antigen presentation, and the dynamics of how cancer cells evade and suppress the host immune system suggest that previous studies may have used suboptimal antigen targets, vaccine designs, and/or trial designs (including patient populations). In 2015, Melief et al. formulated a list of attributes that cancer vaccines would need to have to be successful [6]. In brief, these attributes stress the importance of broad stimulation of CTLs and T helper cells through two mechanisms: (1) selection of appropriate antigens that induce both T cell populations and (2) rational vaccine designs that achieve concentrated delivery of tumor antigens to activated DCs, where epitopes derived from exogenous tumor antigens can be loaded onto both MHC class I (through the cross-presentation pathway) and MHC class II molecules to stimulate CTLs and T helper cells, respectively (Fig. 2) [6, 78]. Over the last decade, therapeutic cancer vaccine strategies have improved, incorporating better immunogenicity, antigen selection, and structural design to meet these criteria.

Copyright Clearance Center, Inc.] CD cluster of differentiation, CLIP class II-associated invariant chain peptide, DC dendritic cell, ER endoplasmic reticulum, FasL Fas ligand, IL interleukin, MHC major histocompatibility complex, SLP synthetic long peptide, TAP transporter of antigen processing, TCR T cell receptor, TNF tumor necrosis factor, TRAIL TNF-related apoptosis-inducing ligand
Optimal antigen processing and presentation by DCs is important for effective immune-mediated tumor cell destruction [6]. Antigens enter DCs through multiple mechanisms, including endocytosis, phagocytosis, pinocytosis, and receptor-mediated uptake. These antigens are processed by DCs into peptide fragments (epitopes) before being loaded onto MHC class I molecules through cross priming or MHC class II molecules through the classical exogenous presentation pathway. T cell recognition of these epitopes occurs via binding between the TCR and the peptide-MHC complex on the DC. Following epitope recognition, CD40L expressed by CD4+ T cells activates DC-expressed CD40 to promote DC maturation and IL-12 secretion. This subsequently stimulates CD28 signaling and activation of CD8+ T cells. When the TCR of an effector CD8+ T cell binds to a tumor cell, an immunological synapse forms and lytic granules are secreted by the effector CD8+ T cell, resulting in tumor cell destruction. Note: Cytosolic and vacuolar pathways for cross-presentation have been described. The figure presents the cytosolic pathway as it has been suggested this is the predominant pathway for cross-presentation [78]. [Figure adapted with permission of the Journal of Clinical Investigation, from “Therapeutic Cancer Vaccines,” Cornelis J.M. Melief et al, Volume 125, Issue 9, 2015; permission conveyed through
3.1 Selecting the Appropriate Antigen
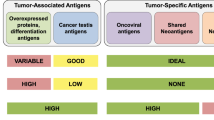
Choosing optimal antigens has been described as the most important consideration in the design of therapeutic vaccines [11]. Antigen selection affects critical vaccine properties, including the ability to generate a strong and broad immune response, target cancer stem cells to prevent relapse, and avoid off-target effects on normal cells. Optimal antigen discovery is hindered by the limited number of suitable immunogenic antigens fitting these criteria within the context of an immense number of potential antigens [14]. Current strategies aim to efficiently identify appropriate antigens for cancer vaccines either in the form of “ideal” shared tumor antigens or more personalized neoantigens.
Shared TAAs are self-proteins with preferential or abnormal expression in cancer cells versus normal cells [11], and these have been the primary type of antigen tested in clinical trials [11]. An important advance in the field occurred in 2009 when a National Cancer Institute (NCI) Pilot Project developed a list of nine “ideal” cancer antigen criteria, which comprised therapeutic function, immunogenicity, stem cell expression, specificity, oncogenicity, expression level and percentage of antigen-positive cells, number of patients with antigen-positive cancers, number of epitopes, and cellular location of expression [14]. The NCI project then used a mathematical model to quantitatively rank 75 antigens according to how well they met these criteria [14]. As shown in Table 1, antigens used in past pivotal trials of cancer vaccines were often not ranked highly on the NCI list, indicating that early vaccines may have lacked efficacy because of suboptimal antigen selection. Tumor antigens employed in these vaccines may also have been insufficiently immunogenic. Several peptide-based vaccines with negative phase 3 results targeted tumor antigens that would have been retrospectively deemed not a high priority target by consensus NCI criteria [14]. For example, the GV1001 peptide vaccine targeted a TAA (human telomerase reverse transcriptase [hTERT]) ranked 23 out of 75 possible antigens by the NCI list [14]. This vaccine failed to generate sufficient immune responses and did not improve OS compared with chemotherapy alone in patients with pancreatic cancer [49].
Development of the NCI’s prioritized list provides the impetus to investigate highly ranked TAAs. Within this list, Wilms’ tumor 1 (WT1) protein, a zinc finger transcription factor, was considered the most encouraging antigen among the 75 assessed [14]. Early-stage trials of cancer vaccines targeting WT1 or other highly ranked TAAs, such as mucin 1 (MUC1) and human epidermal growth factor receptor 2 (HER2)/neu, are ongoing and have demonstrated tumor-specific immune responses [79,80,81,82,83,84,85,86,87,88,89,90,91,92,93]. Notably, randomized phase 2 studies of WT1 vaccines have shown trends toward survival benefit [84, 94], and a phase 3 study of a WT1-based vaccine is being planned (Table 3) [95]. Additionally, the phase 3 ATALANTE-1 trial (NCT02654587) comparing a cancer vaccine targeting five TAAs (angiotensin-converting enzyme [ACE], HER2, melanoma-associated antigen (MAGE) 2, MAGE3, and p53) with chemotherapy in patients with advanced non-small-cell lung cancer who relapsed after checkpoint inhibitor therapy demonstrated an encouraging 1-year OS rate of 46% in the vaccine group (n = 61) compared with 36% in the chemotherapy group (n = 31) [96]. Taken together, these late-stage studies underscore the potential and maturity of targeting highly ranked TAAs as a treatment strategy for cancer.
A limitation of shared TAAs is that they are autologous antigens, and immune self-tolerance mechanisms may deplete or eliminate TAA-specific T cells with high functional avidity, resulting in a tolerized T cell repertoire with relatively low reactivity toward the TAA [6]. Although increased immunogenicity has enabled TAA-based vaccines to break this tolerance, parallel advances in genomics have now allowed the efficient identification of another class of cancer antigen, termed neoantigen, which is not subject to immune self-tolerance mechanisms [6]. Neoantigens are aberrant peptides that arise from genetic and epigenetic alterations (point mutations, insertions/deletions, gene fusions/translocations, splice variants, and post-translational modifications) in cancer cells [97]. Because these alterations are not part of the normal exome or transcriptome, the encoded neoantigens are tumor specific. Mutated peptides that are dissimilar to the self-proteome are more likely to be seen as novel by the immune system, and therefore be more immunogenic, compared with those that are similar to the self-proteome [98].
Broadly, candidate neoantigens are identified through a two-step process. First, whole exome and transcriptome sequencing of normal and cancerous cells allows identification of tumor-specific mutations (i.e., the mutanome) [100]. Next, neoepitopes are prioritized by in silico prediction of the binding affinity of each mutant peptide to MHC molecules. Advances in massive parallel sequencing have drastically accelerated this process, enabling feasible and high-throughput identification of tumor neoantigens for cancer vaccines [99, 100]. Because of these technological advancements and parallel innovations in cancer immunotherapy, the Human Vaccines Project (a public–private partnership with a goal of accelerating the development of cancer vaccines) ranked neoantigens as a high priority target for clinical translation [100].
Several early-stage clinical studies in patients with solid tumors showed that personalized neoantigen vaccines are safe, feasible, and able to augment neoantigen-specific T cell responses [101,102,103,104,105,106,107,108]. Notably, although most neoantigen vaccine clinical studies have been restricted to cancer types with high mutation burdens (e.g., melanoma) and thus more neoantigen potential, recent data from patients with glioblastoma also show neoantigen immune responses for cold tumors with low mutation burdens [106, 109]. Whether the generation of immune responses to neoantigens is therapeutically relevant remains uncertain, as mutations in expressed genes rarely result in the presentation of T cell targetable neoantigens on the cell surface [110] and few neoantigen vaccine studies have attempted to verify that the mutated peptide is present on the surface of the tumor cell. One of the few studies to assess the presence of mutated peptides on the tumor cell surface found 643 genomic mutations among 15 patients with glioblastoma but did not identify any of these mutations in the HLA peptidome by mass spectrometry [111]. Although this study found that neoepitopes induced T cell responses [111], the failure to identify surface mutated peptides calls into question the role of neoantigens for tumors with low tumor mutation burden. Overall, these initial studies of neoantigen cancer vaccines have provided proof of concept, have provided rationale for larger studies and continued research and development [11], and have highlighted a need for neoantigen vaccine studies to verify the presence of targetable, mutated peptides on the tumor cell surface.
There are several ongoing efforts to optimize neoantigen vaccines. Cost-effective and efficient workflows and algorithms for more accurate prediction of which mutated peptides will stimulate the most potent anti-tumor T cell response are being investigated [112, 113]. Innovative neoantigen vaccine designs are also being explored, with preclinical data showing that DNA-based neoantigen vaccines can generate robust CTL-driven anti-tumor responses and delay tumor progression [114]. Furthermore, results from a combined approach using both TAAs and neoantigens in patients with newly diagnosed glioblastoma suggest that mixtures of antigenic targets may provide sustained anti-tumor responses by central memory CTLs and T helper cells [111].
3.2 Evolution of Therapeutic Cancer Vaccine Designs
Over the past decade, vaccine designs have evolved to elicit effective immune responses characterized by potent and broad stimulation of CTLs and T helper cells as well as enhanced antigen presentation by activated DCs. These optimizations, summarized below for the most encouraging platforms, are currently being used in the next generation of therapeutic cancer vaccines with the hope they will lead to improved immune and clinical responses compared with historical experience.
3.2.1 Peptide Vaccines
In response to the observation that short peptides and long protein sequences resulted in inadequate clinical activity (Table 1), extension of the amino acid sequence beyond the minimal-length CTL epitope or other short sequences has been shown to achieve more concentrated and selective delivery of antigens to DCs with sustained presentation [63]. Vaccination with synthetic long peptides has induced more robust and durable T cell responses compared with the minimal-length epitopes in preclinical models [6, 115, 116]. Because of these advantages, modern synthetic peptide vaccine designs commonly use at least one long peptide [6, 117], representing an important advance over the use of minimal-length peptide constructs.
Amino acid substitutions on the native TAA sequence (epitope enhancement) can be rationally implemented to improve binding stability to APCs and thus increase the likelihood of successful antigen presentation to T cells [60, 61, 87]. Modification of the peptide structure to increase amphiphilicity may also increase peptide immunogenicity, as demonstrated in the development of the BiVax peptide/polyinosinic-polycytidylic acid (poly I:C) subunit vaccine [118].
Recent evidence demonstrates that T helper cells play critical roles in induction of strong and long-lasting immune-mediated anti-tumor responses [119, 120]. In particular, targeting T helper type 1 cells is thought to be optimal, as they have been shown to have potent effects in inducing and maintaining anti-tumor immunity, whereas T helper type 2 cells may actually promote neoplastic transformation in certain contexts [121]. While early peptide vaccine designs only targeted CTLs, modern synthetic peptide vaccines now typically include CTL and helper peptides in an effort to increase immunogenicity and improve clinical efficacy [85, 87].
3.2.2 DC Vaccines
The evolution of DC cancer vaccines was sparked by increased insight into DC biology and technology advances, recently reviewed in 2017 by Garg et al., who classified the development of this DC vaccine platform according to first-generation, second-generation, and next-generation designs [122]. Notably, sipuleucel-T, the only approved DC-based vaccine (licensed in 2010), was considered by Garg et al. to be on the borderline of first- and second-generation designs.
In brief, first-generation designs were characterized by the use of immature monocyte-derived DCs; the development of maturation cocktails enabled the consistent use of mature monocyte-derived DCs in second-generation constructs [122]. This advancement was important because compared with immature DCs, mature DCs express higher levels of MHC and costimulatory molecules, produce more cytokines, and traffic more efficiently to lymph nodes [123]. All these effects make mature DCs more potent activators of the immune system, which in turn has been found to translate to improved efficacy in clinical trials. Indeed, Garg et al. reported that in many trials, second-generation DC vaccines produced higher response rates and increased median OS compared with first-generation designs [122].
The transition from second-generation to next-generation DC vaccines was characterized by the use of patient-derived specific DC subsets (e.g., myeloid and plasmacytoid DCs) with specialized functionalities (antigen presentation, interferon responses, migratory capacity) superior to those of monocyte-derived DCs [122, 124]. This transition was enabled by incorporation of antibody-coated magnetic bead technology for more rapid and pure isolation of native DCs compared with older techniques such as density centrifugation [122, 124]. Next-generation DC vaccines are currently being tested in clinical trials [124]; results presented so far from phase 1 or 2 studies have demonstrated their feasibility and safety, with some encouraging OS durations observed [124,125,126,127]. A phase 3 study (NCT02993315) comparing next-generation DC vaccination with placebo as adjuvant therapy for patients with stage III melanoma will provide more robust survival data and clarify the clinical efficacy of this vaccination approach [124].
More recently, a strategy employing intratumoral DCs as part of an in situ vaccine has been described. This in situ vaccine approach uses a triplet consisting of injection of FMS-like tyrosine kinase 3 ligand at the target lesion to generate accumulation of intratumoral DCs, local tumor irradiation to load the intratumoral DCs with TAAs released from dying tumor cells, and injections of a Toll-like receptor (TLR) agonist at the target lesion to drive intratumoral DC activation [128]. In essence, this triplet creates a DC-based vaccine at the site of the tumor [128]. This in situ vaccine was recently evaluated in a phase 1 clinical trial (NCT01976585) for patients with advanced stage indolent non-Hodgkin lymphoma, where it was reported to be well tolerated and capable of producing durable regressions at distant tumor sites via an abscopal effect [128]. Of 11 patients who received the in situ vaccine, eight had partial or complete remissions of the treated tumor with regard to the non-treated tumors, six patients had stable disease or minor regressions lasting 3–18 months, and three achieved remission [128].
3.2.3 Genetic Vaccines
The main limitations of early nucleic acid–based vaccine designs have been limited uptake of the nucleic acid by DCs and other cells, either because of low transfection efficiency or degradation [11], and the resultant low immunogenicity observed in clinical trials. For DNA- or RNA-based vaccines, several upgrades have allowed the prospect of improved transfection rates and immunogenicity as described by Hollingsworth and Jansen [11] including use of electroporation, sonoporation, nanoparticles [129], gene guns, microneedle arrays, needle-free injection [130], and liposomal encapsulation.
Based on encouraging phase 2b data showing significantly higher regression rates of cervical intraepithelial neoplasia compared with placebo [11, 131], a DNA-based vaccine using electroporation is currently being evaluated in two phase 3 studies (REVEAL 1, NCT03185013; REVEAL 2, NCT03721978) to treat patients with precancerous lesions of the cervix (high-grade squamous intraepithelial lesions associated with human papillomavirus). Early-stage studies are evaluating DNA-based vaccines using electroporation for a variety of solid tumors (NCT03199040, NCT03122106, NCT03532217, NCT03439085, NCT02204098, and NCT04397003). In phase 1 studies, RNA-based vaccines using either electroporation [108] or RNA-lipoplexes [132] to improve systemic DC targeting have demonstrated encouraging immune-mediated anti-tumor activity for patients with melanoma.
Other methods to improve the immunogenicity of nucleic acid–based vaccines have been recently reviewed by Lopes et al. [133]. To break immune tolerance and target multiple TAAs, DNA vaccines encoding xenoantigens or chimeric proteins have been studied [133]. In animal models, chimeric DNA vaccines have induced potential anti-tumor effects [134, 135], and one such vaccine is approved for the treatment of canine melanoma. Numerous human clinical trials are currently evaluating polyepitope DNA vaccines, which aim to induce a broad T cell response through the simultaneous delivery of multiple antigens [133].
3.3 The Role of Adjuvants
Effective therapeutic cancer vaccines rely on antigen presentation and activation of the immune system by DCs; however, suppression of DC maturation and function is a hallmark of cancer immune evasion [136]. Even worse, many subsets of DCs are in an immature state and produce “self”-tolerizing messages to the immune system. Therefore, cancer vaccines without DC activators may actually convey a tolerizing signal to the immune system and diminish endogenous immune response [137]. Thus, activation of DCs with immunostimulatory adjuvants is a critical component of many cancer vaccine strategies [136].
An advance in the design of cancer vaccines has been the inclusion of adjuvants that can trigger pattern recognition receptors, such as TLRs, Nod-like receptors (NLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), stimulator of interferon genes (STING), and CD40 agonists, to signal the immune system that the vaccine antigen is both foreign and dangerous [2, 6, 11, 138]. This is especially important for peptide vaccine platforms, because unlike microbe- or nucleic acid–based platforms, peptide antigens do not inherently present danger signals to the immune system [11]. Without this signal, the immune system cannot mount a strong anti-tumor response due to a lack of costimulation and efficient antigen presentation by DCs.
Novel adjuvants, such as TLR agonists, have been tested in preclinical and clinical studies [11], with evidence of potent DC activation and generation of strong T cell responses [139, 140]. An important finding is that co-delivery of the peptide antigen and TLR agonist, either through peptide-agonist conjugation or nanocarriers, results in improved DC targeting, DC activation, and trafficking to draining lymph nodes [141, 142]. Recent peptide vaccine formulations have primarily employed Montanide ISA-51, TLR agonists such as poly I:C or CpG, or immunostimulatory cytokines such as GM-CSF as adjuvants [117]; however, there is currently no consensus about what the optimal adjuvant is for a given peptide vaccine [117], representing a potentially fruitful avenue of research to further optimize vaccine design.
4 Future Perspective
The historical experience with therapeutic cancer vaccines coupled with fundamental advances in understanding of the immunobiology of cancer have provided a road map for future vaccine development. The key challenges that must be overcome are identifying antigens and vaccine vectors that will lead to strong and broad T cell responses, tailoring vaccine designs to achieve optimal antigen presentation by professional APCs, and finding combination partners employing complementary mechanisms of action to overcome the diverse methods that cancer cells use to evade and suppress the immune system [11]. In recent years, the field has risen to meet this challenge, with many encouraging upgrades to antigen selection and vaccine designs. Combination strategies with a variety of other agents, including immunotherapies, chemotherapies, and radiotherapy, have also been investigated in preclinical and clinical studies [11]. These refinements will need to be validated in appropriately designed, randomized, phase 3 studies. Consequently, despite decades of lackluster progress, therapeutic cancer vaccines are now primed to emerge as central components of cancer therapy due to these advancements in biology and technology.
Therapeutic cancer vaccines may fill a niche not currently met by conventional therapies or other immunotherapies. Clinical experience suggests that vaccines are safe and can elicit long-term immune memory responses important for durable disease control [11]. This experience coupled with the existence of multiple mechanisms of immunosuppression in advanced disease suggest that vaccines may be particularly well-suited early in the course of the disease or in the minimal residual disease setting. Indeed, when there has been apparent benefit from cancer vaccines, it has been in the minimal residual disease setting [143].
Another potential role for vaccination is to augment lost immunity to oncogenic proteins where immunity is lost through oncogenesis [144, 145]. Cancer vaccines could also potentially be used to prevent disease through the targeting of antigens whose upregulation is associated with resistance to therapy [146].
Therapeutic cancer vaccines may also help actualize the full potential of immunotherapies that have already had an impact in the clinic [12]. For example, it is known that although checkpoint inhibitors are effective for “hot” tumors characterized by infiltration of primed and active T cells, they lack potency for “cold” tumors that do not have these immune cells. By priming tumor-specific T cells and mobilizing them to the tumor, essentially turning “cold” tumors “hot,” vaccine therapies restore the ability of checkpoint inhibitors to unleash T cell-mediated tumor destruction [12, 128]. Similarly, whereas chimeric antigen receptor (CAR)-T cell therapy is now an established therapy for certain hematologic malignancies, demonstrating efficacy in solid tumors has been difficult. A recent preclinical study showed that injection of a vaccine consisting of amphiphile CAR-T ligands primed CAR-T cells and enhanced their efficacy in solid tumor models [147].
There are several encouraging avenues to further improve the efficacy of therapeutic cancer vaccines. One currently being investigated is the use of heterologous prime boosting whereby a TAA is first delivered by a specific vector during a priming vaccination and then subsequently delivered by a different vector during later boosting vaccinations [11]. This strategy overcomes immune-mediated inactivation of the initial viral vector, allowing repeated vaccination against the TAA target and potentially enhanced immunogenicity [11]. Another approach to improve the efficacy of cancer vaccines is the development of strategies to induce antibody responses with anti-tumor activity. For example, one study found that multi-site injections enhanced the number of TAA-specific antibodies compared with single bolus injections [148].
Finally, it is anticipated that cancer vaccines will benefit from advances in the speed, cost, and efficiency of molecular sequencing, artificial intelligence, and cellular engineering. These techniques may enable the quick and complete interrogation of the immune response (changes in immune milieu, tumor immune escape mechanisms) to a cancer vaccine, allowing subsequent vaccines to be tailored based on this response [149].
References
Butterfield LH. Cancer vaccines. BMJ. 2015;350:h988.
Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8(5):351–60.
Hoption Cann SA, van Netten JP, van Netten C. Dr William Coley and tumour regression: a place in history or in the future. Postgrad Med J. 2003;79(938):672–80.
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8.
Ott PA, Wu CJ. Cancer vaccines: steering T cells down the right path to eradicate tumors. Cancer Discov. 2019;9(4):476–81.
Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Investig. 2015;125(9):3401–12.
Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines combined with immune checkpoint antibodies promote cytotoxic T-cell activity and tumor eradication. Cancer Immunol Res. 2016;4(2):95–100.
Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38(1):1–11.
Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother. 2011;34(5):409–18.
Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight. 2018;3(20):e122857.
Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vacc. 2019;4:7.
Maeng HM, Berzofsky JA. Strategies for developing and optimizing cancer vaccines. F1000Res. 2019;8:F1000 Faculty Rev-654.
DeMaria PJ, Bilusic M. Cancer vaccines. Hematol Oncol Clin N Am. 2019;33:199–214.
Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–37.
Madan RA, Bilusic M, Heery C, Schlom J, Gulley JL. Clinical evaluation of TRICOM vector therapeutic cancer vaccines. Semin Oncol. 2012;39(3):296–304.
Gulley JL, Borre M, Vogelzang NJ, Ng S, Agarwal N, Parker CC, et al. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37(13):1051–61.
Chawla SP, Tine BAV, Pollack S, Ganjoo KN, Elias AD, Riedel RF, et al. A phase II randomized study of CMB305 and atezolizumab versus atezolizumab in NY-ESO-1+ soft tissue sarcoma: Analysis of immunogenicity, tumor control, and patient survival. J Clin Oncol. 2019;37(15_suppl):11011.
Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhasz E, Ramlau R, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015;51(16):2321–9.
Vishnu P, Tan WW. Update on options for treatment of metastatic castration-resistant prostate cancer. Onco Targets Ther. 2010;3:39–51.
Morton DL, Mozzillo N, Thompson JF, Kelley MC, Faries M, Wagner J, et al. An international, randomized, phase III trial of bacillus Calmette-Guerin (BCG) plus allogeneic melanoma vaccine (MCV) or placebo after complete resection of melanoma metastatic to regional or distant sites. J Clin Oncol. 2007;25(18_suppl):8508.
Ogi C, Aruga A. Clinical evaluation of therapeutic cancer vaccines. Hum Vacc Immunother. 2013;9(5):1049–57.
Faries MB, Mozzillo N, Kashani-Sabet M, Thompson JF, Kelley MC, DeConti RC, et al. Long-term survival after complete surgical resection and adjuvant immunotherapy for distant melanoma metastases. Ann Surg Oncol. 2017;24(13):3991–4000.
Sondak VK, Liu PY, Tuthill RJ, Kempf RA, Unger JM, Sosman JA, et al. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: overall results of a randomized trial of the Southwest Oncology Group. J Clin Oncol. 2002;20(8):2058–66.
von Eschen KB, Mitchell MS. Phase III trial of melacine® melanoma theraccine versus combination chemotherapy in the treatment of stage IV melanoma: 179. Melanoma Res. 1997;7:S51.
NewLink Genetics Announces Results from Phase 3 IMPRESS Trial of Algenpantucel-L for Patients with Resected Pancreatic Cancer. NewLink Genetics Corporation press release, May 9, 2016. https://www.globenewswire.com/news-release/2016/05/09/837878/0/en/NewLink-Genetics-Announces-Results-from-Phase-3-IMPRESS-Trial-of-Algenpantucel-L-for-Patients-with-Resected-Pancreatic-Cancer.html. Accessed 6 June 2020.
Hersey P, Coates AS, McCarthy WH, Thompson JF, Sillar RW, McLeod R, et al. Adjuvant immunotherapy of patients with high-risk melanoma using vaccinia viral lysates of melanoma: results of a randomized trial. J Clin Oncol. 2002;20(20):4181–90.
Wallack MK, Sivanandham M, Balch CM, Urist MM, Bland KI, Murray D, et al. A phase III randomized, double-blind multiinstitutional trial of vaccinia melanoma oncolysate-active specific immunotherapy for patients with stage II melanoma. Cancer. 1995;75(1):34–42.
Wallack MK, Sivanandham M, Balch CM, Urist MM, Bland KI, Murray D, et al. Surgical adjuvant active specific immunotherapy for patients with stage III melanoma: the final analysis of data from a phase III, randomized, double-blind, multicenter vaccinia melanoma oncolysate trial. J Am Coll Surg. 1998;187(1):69–77.
Suriano R, Rajoria S, George AL, Geliebter J, Tiwari RK, Wallack M. Follow-up analysis of a randomized phase III immunotherapeutic clinical trial on melanoma. Mol Clin Oncol. 2013;1(3):466–72.
Figlin R, Nicolette C, Tannir N, Tykodi S, Chen D, Master V, et al. Interim analysis of the phase 3 ADAPT trial evaluating rocapuldencel-T (AGS-003), an individualized immunotherapy for the treatment of newly-diagnosed patients with metastatic renal cell carcinoma (mRCC). Ann Oncol. 2017;28(suppl_5): 1137O.
Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17(4):563–70.
Leach B. OncLive. Allovectin falters in late-stage melanoma trial. Published August 13, 2013. https://www.onclive.com/view/allovectin-falters-in-late-stage-melanoma-trial. Accessed 24 June 2020.
Bedikian AY, Del Vecchio M. Allovectin-7 therapy in metastatic melanoma. Expert Opin Biol Ther. 2008;8(6):839–44.
Powell K. DNA vaccines–back in the saddle again? Nat Biotechnol. 2004;22(7):799–801.
Eggermont AM, Suciu S, Rutkowski P, Marsden J, Santinami M, Corrie P, et al. Adjuvant ganglioside GM2-KLH/QS-21 vaccination versus observation after resection of primary tumor > 1.5 mm in patients with stage II melanoma: results of the EORTC 18961 randomized phase III trial. J Clin Oncol. 2013;31(30):3831–7.
Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK, Agarwala SS, Ernstoff MS, et al. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol. 2001;19(9):2370–80.
Sabbatini P, Harter P, Scambia G, Sehouli J, Meier W, Wimberger P, et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: a phase III trial of the AGO OVAR, COGI, GINECO, and GEICO—the MIMOSA study. J Clin Oncol. 2013;31(12):1554–61.
Giaccone G, Debruyne C, Felip E, Chapman PB, Grant SC, Millward M, et al. Phase III study of adjuvant vaccination with Bec2/bacille Calmette-Guerin in responding patients with limited-disease small-cell lung cancer (European Organisation for Research and Treatment of Cancer 08971–08971B; Silva Study). J Clin Oncol. 2005;23(28):6854–64.
Levy R, Ganjoo KN, Leonard JP, Vose JM, Flinn IW, Ambinder RF, et al. Active idiotypic vaccination versus control immunotherapy for follicular lymphoma. J Clin Oncol. 2014;32(17):1797–803.
Freedman A, Neelapu SS, Nichols C, Robertson MJ, Djulbegovic B, Winter JN, et al. Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte-macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J Clin Oncol. 2009;27(18):3036–43.
Schuster SJ, Neelapu SS, Gause BL, Janik JE, Muggia FM, Gockerman JP, et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J Clin Oncol. 2011;29(20):2787–94.
Miles D, Roche H, Martin M, Perren TJ, Cameron DA, Glaspy J, et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist. 2011;16(8):1092–100.
Dreno B, Thompson JF, Smithers BM, Santinami M, Jouary T, Gutzmer R, et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19(7):916–29.
Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(6):822–35.
Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(1):59–68.
Mitchell P, Thatcher N, Socinski MA, Wasilewska-Tesluk E, Horwood K, Szczesna A, et al. Tecemotide in unresectable stage III non-small-cell lung cancer in the phase III START study: updated overall survival and biomarker analyses. Ann Oncol. 2015;26(6):1134–42.
Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, Buonaguro L. Antigen-specific vaccines for cancer treatment. Hum Vacc Immunother. 2014;10(11):3332–46.
Xia W, Wang J, Xu Y, Jiang F, Xu L. L-BLP25 as a peptide vaccine therapy in non-small cell lung cancer: a review. J Thorac Dis. 2014;6(10):1513–20.
Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15(8):829–40.
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–85.
Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19(4):713–23.
Yamaue H, Tsunoda T, Tani M, Miyazawa M, Yamao K, Mizuno N, et al. Randomized phase II/III clinical trial of elpamotide for patients with advanced pancreatic cancer: PEGASUS-PC Study. Cancer Sci. 2015;106(7):883–90.
Lawson DH, Lee S, Zhao F, Tarhini AA, Margolin KA, Ernstoff MS, et al. Randomized, placebo-controlled, phase III trial of yeast-derived granulocyte-macrophage colony-stimulating factor (gm-csf) versus peptide vaccination versus gm-csf plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV melanoma: a trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). J Clin Oncol. 2015;33(34):4066–76.
Mittendorf EA, Lu B, Melisko M, Price Hiller J, Bondarenko I, Brunt AM, et al. Efficacy and safety analysis of nelipepimut-s vaccine to prevent breast cancer recurrence: a randomized, multicenter, phase III clinical trial. Clin Cancer Res. 2019;25(14):4248–54.
Clifton GT, Peoples GE, Mittendorf EA. The development and use of the E75 (HER2 369–377) peptide vaccine. Future Oncol. 2016;12(11):1321–9.
Wood C, Srivastava P, Bukowski R, Lacombe L, Gorelov AI, Gorelov S, et al. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet. 2008;372(9633):145–54.
Testori A, Richards J, Whitman E, Mann GB, Lutzky J, Camacho L, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol. 2008;26(6):955–62.
Bloch O, Shi Q, Anderson SK, Knopp M, Raizer J, Clarke J, et al. ATIM-14. ALLIANCE A071101: a phase II randomized trial comparing the efficacy of heat shock protein peptide COMPLEX-96 (HSPPC-96) vaccine given with bevacizumab versus bevacizumab alone in the treatment of surgically resectable recurrent glioblastoma. Neuro-Oncology. 2017;19(suppl_6):vi29.
Forster V. Cancer Therapy Advisor. Phase 3 trial for oncolytic viral therapy pexa-vec in advanced liver cancer terminated early. 2019. https://www.cancertherapyadvisor.com/home/cancer-topics/general-oncology/phase-3-trial-for-oncolytic-viral-therapy-pexa-vec-in-advanced-liver-cancer-terminated-early/2/. Accessed 24 June 2020.
Kumai T, Fan A, Harabuchi Y, Celis E. Cancer immunotherapy: moving forward with peptide T cell vaccines. Curr Opin Immunol. 2017;47:57–63.
Kumai T, Kobayashi H, Harabuchi Y, Celis E. Peptide vaccines in cancer-old concept revisited. Curr Opin Immunol. 2017;45:1–7.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9.
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38(4):1033–42.
Faure F, Mantegazza A, Sadaka C, Sedlik C, Jotereau F, Amigorena S. Long-lasting cross-presentation of tumor antigen in human DC. Eur J Immunol. 2009;39(2):380–90.
Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur J Immunol. 2013;43(10):2554–65.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
McKay RR, Hafron JM, Ferro C, Wilfehrt HM, Fitch K, Flanders SC, et al. A retrospective observational analysis of overall survival with sipuleucel-T in Medicare beneficiaries treated for advanced prostate cancer. Adv Ther. 2020;37(12):4910–29.
Huang X, Ye D, Thorpe PE. Enhancing the potency of a whole-cell breast cancer vaccine in mice with an antibody-IL-2 immunocytokine that targets exposed phosphatidylserine. Vaccine. 2011;29(29–30):4785–93.
Graner MW, Likhacheva A, Davis J, Raymond A, Brandenberger J, Romanoski A, et al. Cargo from tumor-expressed albumin inhibits T-cell activation and responses. Cancer Res. 2004;64(21):8085–92.
Aleynick M, Svensson-Arvelund J, Flowers CR, Marabelle A, Brody JD. Pathogen molecular pattern receptor agonists: treating cancer by mimicking infection. Clin Cancer Res. 2019;25(21):6283–94.
Kantoff PW, Gulley JL, Pico-Navarro C. Revised overall survival analysis of a phase II, randomized, double-blind, controlled study of PROSTVAC in men with metastatic castration-resistant prostate cancer. J Clin Oncol. 2017;35(1):124–5.
Klinman DM, Yamshchikov G, Ishigatsubo Y. Contribution of CpG motifs to the immunogenicity of DNA vaccines. J Immunol. 1997;158(8):3635–9.
Antonelli AC, Binyamin A, Hohl TM, Glickman MS, Redelman-Sidi G. Bacterial immunotherapy for cancer induces CD4-dependent tumor-specific immunity through tumor-intrinsic interferon-γ signaling. Proc Natl Acad Sci USA. 2020;117(31):18627–37.
Toda M, Martuza RL, Rabkin SD. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol Ther. 2000;2(4):324–9.
Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303.
Kaufman HL, Kim DW, DeRaffele G, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–30.
Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–69.
Yanagisawa R, Koizumi T, Koya T, Sano K, Koido S, Nagai K, et al. WT1-pulsed dendritic cell vaccine combined with chemotherapy for resected pancreatic cancer in a phase I study. Anticancer Res. 2018;38(4):2217–25.
Crosby EJ, Gwin W, Blackwell K, Marcom PK, Chang S, Maecker HT, et al. Vaccine-induced memory CD8+ T cells provide clinical benefit in HER2 expressing breast cancer: a mouse to human translational study. Clin Cancer Res. 2019;25(9):2725–36.
Ueda Y, Ogura M, Miyakoshi S, Suzuki T, Heike Y, Tagashira S, et al. Phase 1/2 study of the WT1 peptide cancer vaccine WT4869 in patients with myelodysplastic syndrome. Cancer Sci. 2017;108(12):2445–53.
Tsuboi A, Hashimoto N, Fujiki F, Morimoto S, Kagawa N, Nakajima H, et al. A phase I clinical study of a cocktail vaccine of Wilms’ tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol Immunother. 2019;68(2):331–40.
Ogasawara M, Miyashita M, Yamagishi Y, Ota S. Phase I/II pilot study of Wilms’ tumor 1 peptide-pulsed dendritic cell vaccination combined with conventional chemotherapy in patients with head and neck cancer. Ther Apher Dial. 2019;23(3):279–88.
Nishida S, Ishikawa T, Egawa S, Koido S, Yanagimoto H, Ishii J, et al. Combination gemcitabine and WT1 peptide vaccination improves progression-free survival in advanced pancreatic ductal adenocarcinoma: a phase II randomized study. Cancer Immunol Res. 2018;6(3):320–31.
Miyakoshi S, Usuki K, Matsumura I, Ueda Y, Iwasaki H, Miyamoto T, et al. Preliminary results from a phase 1/2 study of DSP-7888, a novel WT1 peptide-based vaccine, in patients with myelodysplastic syndrome (MDS). Blood. 2016;128(22):4335.
Matsuda T, Takeuchi H, Sakurai T, Mayanagi S, Booka E, Fujita T, et al. Pilot study of WT1 peptide-pulsed dendritic cell vaccination with docetaxel in esophageal cancer. Oncol Lett. 2018;16(1):1348–56.
Maslak PG, Dao T, Bernal Y, Chanel SM, Zhang R, Frattini M, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018;2(3):224–34.
Katsuda M, Miyazawa M, Ojima T, Katanuma A, Hakamada K, Sudo K, et al. A double-blind randomized comparative clinical trial to evaluate the safety and efficacy of dendritic cell vaccine loaded with WT1 peptides (TLP0-001) in combination with S-1 in patients with advanced pancreatic cancer refractory to standard chemotherapy. Trials. 2019;20(1):242.
Hirabayashi K, Yanagisawa R, Saito S, Higuchi Y, Koya T, Sano K, et al. Feasibility and immune response of WT1 peptide vaccination in combination with OK-432 for paediatric solid tumors. Anticancer Res. 2018;38(4):2227–34.
Hanada S, Tsuruta T, Haraguchi K, Okamoto M, Sugiyama H, Koido S. Long-term survival of pancreatic cancer patients treated with multimodal therapy combined with WT1-targeted dendritic cell vaccines. Hum Vaccin Immunother. 2019;15(2):397–406.
De Groot JF, Cloughesy TF, Pitz MW, Narita Y, Nonomura T. A randomized, multicenter phase 2 study of DSP-7888 dosing emulsion in combination with bevacizumab (Bev) versus Bev alone in patients with recurrent or progressive glioblastoma. J Clin Oncol. 2018;36(15_suppl):TPS2071.
Fu S, Piccioni DE, Liu H, Vincas Lucas R, Aregawi D, Yamaguchi K, et al. Initial phase 1 study of WT2725 dosing emulsion in patients with advanced malignancies. J Clin Oncol. 2017;35(15_suppl):2066.
Berneman ZN, Anguille S, Willemen Y, Van de Velde A, Germonpre P, Huizing M, et al. Vaccination of cancer patients with dendritic cells electroporated with mRNA encoding the Wilms’ tumor 1 protein (WT1): correlation of clinical effect and overall survival with T-cell response. Cytotherapy. 2019;21(5):S10.
Zauderer MG, Tsao AS, Dao T, Panageas K, Lai WV, Rimner A, et al. A randomized phase II trial of adjuvant galinpepimut-S, WT-1 analogue peptide vaccine, after multimodality therapy for patients with malignant pleural mesothelioma. Clin Cancer Res. 2017;23(24):7483–9.
SELLAS Life Sciences Group. SELLAS Life Sciences announces review of strategic alternatives. 2019. https://www.sellaslifesciences.com/investors/news/News-Details/2019/SELLAS-Life-Sciences-Announces-Review-of-Strategic-Alternatives/default.aspx. Accessed 12 July 2019.
Giaccone G, Felip E, Cobo M, Campelo RG, DENIS F, Quoix E, et al. Activity of OSE-2101 in HLA-A2+ non-small cell lung cancer (NSCLC) patients after failure to immune checkpoint inhibitors (ICI): Step 1 results of phase III ATALANTE-1 randomised trial. Ann Oncol. 2020;31(9):S814–5.
Olweus J, Lund-Johansen F. Finding Neo (antigens, that is). Blood. 2019;134(2):108–9.
Richman LP, Vonderheide RH, Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019;9(4):375-382.e4.
Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Investig. 2015;125(9):3413–21.
Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, et al. The Human Vaccines Project: a roadmap for cancer vaccine development. Sci Transl Med. 2016;8(334):334ps9.
Cafri G, Gartner JJ, Hopson K, Meehan RS, Zaks TZ, Robbins P, et al. Immunogenicity and tolerability of personalized mRNA vaccine mRNA-4650 encoding defined neoantigens expressed by the autologous cancer. J Clin Oncol. 2019;37(15_suppl):2643.
Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–8.
Fang Y, Shou J, Mo F, Wang H, Zhang S, Hongsen L, et al. Clinical study of a personalized neoantigen cancer vaccine in treating patients with advanced malignant tumor. Cancer Res 2019;79(13_Suppl): CT006.
Hecht JR, Goldman JW, Hayes S, Balli D, Princiotta MF, Dennie JG, et al. Safety and immunogenicity of a personalized neoantigen-Listeria vaccine in cancer patients. Cancer Res 2019;79(13_Suppl): CT007.
Hu-Lieskovan S, Ott PA, Naing A, Besada RH, Gates SJ, Kohler VR, et al. The personalized vaccine, NEO-PV-01 with anti-PD1, induces neoantigen-specific de novo immune responses in patients with advanced metastatic melanoma: association with clinical outcomes. Cancer Res 2019;79(13_Suppl): 942.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21.
Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–6.
Johanns TM, Miller CA, Liu CJ, Perrin RJ, Bender D, Kobayashi DK, et al. Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. Oncoimmunology. 2019;8(4):e1561106.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–5.
Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods. 2019;16(1):63–6.
Tran NH, Qiao R, Xin L, Chen X, Shan B, Li M. Personalized deep learning of individual immunopeptidomes to identify neoantigens for cancer vaccines. Nat Mach Intell. 2020;2:764–71.
Duperret EK, Perales-Puchalt A, Stoltz R, Mandloi N, Barlow J, Chaudhuri A, et al. A synthetic DNA, multi-neoantigen vaccine drives predominately MHC class I CD8+ T-cell responses, impacting tumor challenge. Cancer Immunol Res. 2019;7(2):174–82.
Zwaveling S, Ferreira Mota SC, Nouta J, Johnson M, Lipford GB, Offringa R, et al. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169(1):350–8.
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179(8):5033–40.
Calvo Tardon M, Allard M, Dutoit V, Dietrich PY, Walker PR. Peptides as cancer vaccines. Curr Opin Pharmacol. 2019;47:20–6.
Cho HI, Barrios K, Lee YR, Linowski AK, Celis E. BiVax: a peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol Immunother. 2013;62(4):787–99.
Kumai T, Lee S, Cho HI, Sultan H, Kobayashi H, Harabuchi Y, et al. Optimization of peptide vaccines to induce robust antitumor CD4 T-cell responses. Cancer Immunol Res. 2017;5(1):72–83.
Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168(3):487-502.e15.
Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. 2014;2(2):91–8.
Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017;38(8):577–93.
Santos PM, Butterfield LH. Dendritic cell-based cancer vaccines. J Immunol. 2018;200(2):443–9.
Bol KF, Schreibelt G, Rabold K, Wculek SK, Schwarze JK, Dzionek A, et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J Immunother Cancer. 2019;7(1):109.
Schreibelt G, Bol KF, Westdorp H, Wimmers F, Aarntzen EH, Duiveman-de Boer T, et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin Cancer Res. 2016;22(9):2155–66.
Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013;73(3):1063–75.
Westdorp H, Oort IMv, Creemers JHA, Schreibelt G, Mehra N, de Goede AL, et al. Myeloid and plasmacytoid dendritic cell vaccinations for castration-resistant prostate cancer patients. J Clin Oncol. 2018;36(6_suppl):219.
Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. 2019;25(5):814–24.
Lim M, Badruddoza AZM, Firdous J, Azad M, Mannan A, Al-Hilal TA, et al. Engineered nanodelivery systems to improve DNA vaccine technologies. Pharmaceutics. 2020;12(1):30.
Teixeira L, Medioni J, Garibal J, Adotevi O, Doucet L, Durey MD, et al. A first-in-human phase I study of INVAC-1, an optimized human telomerase DNA vaccine in patients with advanced solid tumors. Clin Cancer Res. 2020;26(3):588–97.
Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386(10008):2078–88.
Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401.
Lopes A, Vandermeulen G, Preat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38(1):146.
Soong RS, Trieu J, Lee SY, He L, Tsai YC, Wu TC, et al. Xenogeneic human p53 DNA vaccination by electroporation breaks immune tolerance to control murine tumors expressing mouse p53. PLoS ONE. 2013;8(2):e56912.
Grosenbaugh DA, Leard AT, Bergman PJ, Klein MK, Meleo K, Susaneck S, et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res. 2011;72(12):1631–8.
Bowen WS, Svrivastava AK, Batra L, Barsoumian H, Shirwan H. Current challenges for cancer vaccine adjuvant development. Expert Rev Vacc. 2018;17(3):207–15.
Wilson NS, El-Sukkari D, Villadangos JA. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood. 2004;103(6):2187–95.
Bonam SR, Partidos CD, Halmuthur SKM, Muller S. An overview of novel adjuvants designed for improving vaccine efficacy. Trends Pharmacol Sci. 2017;38(9):771–93.
Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007;5:10.
Naumann K, Wehner R, Schwarze A, Petzold C, Schmitz M, Rohayem J. Activation of dendritic cells by the novel Toll-like receptor 3 agonist RGC100. Clin Dev Immunol. 2013;2013:283649.
Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16(4):489–96.
Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol. 2020;38(3):320–32.
Rao B, Han M, Wang L, Gao X, Huang J, Huang M, et al. Clinical outcomes of active specific immunotherapy in advanced colorectal cancer and suspected minimal residual colorectal cancer: a meta-analysis and system review. J Transl Med. 2011;9:17.
Datta J, Fracol M, McMillan MT, Berk E, Xu S, Goodman N, et al. Association of depressed anti-HER2 T-helper type 1 response with recurrence in patients with completely treated HER2-positive breast cancer: role for immune monitoring. JAMA Oncol. 2016;2(2):242–6.
Lowenfeld L, Mick R, Datta J, Xu S, Fitzpatrick E, Fisher CS, et al. Dendritic cell vaccination enhances immune responses and induces regression of HER2pos DCIS independent of route: results of randomized selection design trial. Clin Cancer Res. 2017;23(12):2961–71.
Osada T, Hartman ZC, Wei J, Lei G, Hobeika AC, Gwin WR, et al. Polyfunctional anti-human epidermal growth factor receptor 3 (anti-HER3) antibodies induced by HER3 vaccines have multiple mechanisms of antitumor activity against therapy resistant and triple negative breast cancers. Breast Cancer Res. 2018;20(1):90.
Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365(6449):162–8.
Mould RC, AuYeung AWK, van Vloten JP, Susta L, Mutsaers AJ, Petrik JJ, et al. Enhancing immune responses to cancer vaccines using multi-site injections. Sci Rep. 2017;7(1):8322.
Miho E, Yermanos A, Weber CR, Berger CT, Reddy ST, Greiff V. Computational strategies for dissecting the high-dimensional complexity of adaptive immune repertoires. Front Immunol. 2018;9:224.
Acknowledgements
Medical writing assistance was provided by Andrew Gomes, PhD, of Ashfield Healthcare Communications, a UDG Healthcare plc company, and funded by Sumitomo Dainippon Pharma Oncology, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Medical writing assistance was funded by Sumitomo Dainippon Pharma Oncology, Inc.
Conflict of interest
MAM is a consultant/advisory board member for Roche/Genentech, AstraZeneca/Medimmune, Celldex, Eli Lilly, and Gritstone and has received research funding from Merck, BMS, Etubics, and Alphavax. WRG and DAM declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
All authors were involved in the development of the content and direction of this article. Additionally, all authors revised it critically for important intellectual content; approved the final version; and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Morse, M.A., Gwin, W.R. & Mitchell, D.A. Vaccine Therapies for Cancer: Then and Now. Targ Oncol 16, 121–152 (2021). https://doi.org/10.1007/s11523-020-00788-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-020-00788-w