
Abstract
We studied the effect of two rare mutations (rs144662445 and rs149979685) in the A-kinase anchoring protein 9 (AKAP9) gene, previously associated with Alzheimer disease (AD) in African Americans (AA), on post-translational modifications of AD-related pathogenic molecules, amyloid precursor protein (APP) and microtubule-associated protein Tau using lymphoblastoid cell lines (LCLs) from 11 AA subjects with at least one AKAP9 mutation and 17 AA subjects lacking these mutations. LCLs were transduced by viral vectors expressing causative AD mutations in APP or human full-length wild type Tau. Cell lysates were analyzed for total APP, Aβ40, and total and T181 phospho-Tau (pTau). AKAP9 mutations had no effect on Aβ40/APP, but significantly increased pTau/Tau ratio in LCLs treated with phosphodiesterase-4 inhibitor rolipram, which activates protein kinase A. Proteomic analysis of Tau interactome revealed enrichment of RNA binding proteins and decrease of proteasomal molecules in rolipram-treated cells with AKAP9 mutations. This study shows the impact of rare functional AKAP9 mutations on Tau, a central mechanism of AD pathogenesis, in LCLs derived from AD and control subjects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
We previously identified by whole exome sequencing two rare specific variants in AKAP9 (rs144662445 and rs149979685) in a sample of African American (AA) Alzheimer disease (AD) cases and controls (Logue et al. 2014). The odds of AD were increased 2.75-fold among individuals possessing one or both of these variants (AKAP9+). These SNPs were not present in more than 4000 sequenced individuals with European ancestry (The 1000 Genomes Project Consortium 2010), suggesting that this variant is unique to persons of African ancestry. AKAP9 encodes A kinase-anchoring protein 9 (AKAP9), is ubiquitously expressed in the central nervous system and periphery, and is a scaffold protein that physically interacts with multiple protein kinase-related molecules, such as the regulatory subunit of protein kinase A (PPKAR2A) (Aranda et al. 2010), CDK5RAP2 (Wu et al. 2010), and GSK3B (Varjosalo et al. 2013). AKAP9 has also been associated with long QT syndrome (Chen et al. 2007). However, the functional characterization of AA-specific variants has not been studied.
AKAP9 tethers protein kinase A (PKA) to subcellular loci via binding to the amino-terminal dimerization/docking (D/D) domain of PKA R subunits, and enhances its sensitivity to cAMP (Terrin et al. 2012). Amino acids 2327–2602 of the AKAP9 long isoform AKAP450 bind to PKA RII; a point mutation of amino acid 2566 of this isoform interferes with this interaction (Witczak et al. 1999). The AD-associated rs149979685 variant encodes a S3771 L mutation of the AKAP9 Q99996–1 transcript and is predicted to have a functional consequence. The rs144662445 variant encodes an I2558M mutation of the AKAP9 Q99996–1 transcript and is located within the PKA RII binding site, suggesting a loss-of-function mutation (Logue et al. 2014). It has been reported that PKA is biologically implicated in Tau phosphorylation, which is regulated by the intracellular level of and sensitivity to cyclic AMP (cAMP). PKA promotes the activity of glycogen synthase kinase-3β (GSK3B) and enhances Tau phosphorylation (Liu et al. 2006). The level of cAMP is regulated via its catalysis by phosphodiesterase 4A (PDE4A), which is known to interact with AKAP9 (Terrin et al. 2012). Tau phosphorylation by PKA, and a corresponding loss of PDE4A, confers risk for degeneration in aging association cortex (Carlyle et al. 2014). The role of PKA on amyloid-beta peptide (Aβ) generation from amyloid precursor protein (APP) is poorly understood. Because Tau and Aβ dysregulation are the hallmarks of AD pathogenesis, we hypothesized that AA AD-associated coding mutations in AKAP9 might alter the activation status of PKA and increase phosphorylation of the Tau protein and Aβ production.
Non-brain cell sources have emerged as a powerful alternative for investigating gene expression differences ex vivo in neuropsychiatric disorders including schizophrenia, bipolar disorder and autism using peripheral blood leukocytes (Middleton et al. 2005; Tsuang et al. 2005; Hu et al. 2006) and lymphoblastoid cell lines (LCLs) (Kakiuchi et al. 2003; Vawter et al. 2004). LCLs have also been employed to study gene expression changes related to Aβ (Johnston et al. 1997; Hadar et al. 2016) and TDP-43 (Borroni et al. 2009). Taking into account that LCLs are easier to manipulate and culture than genetically engineered neurons or pluripotent stem cells, while faithfully recapitulating epigenomic signatures (Hadar et al. 2016) and providing material for multiple experiments, we tested the effect of AD-associated AKAP9 missense mutations on Tau and Aβ using 28 LCLs from AD cases and controls with or without the rs144662445 and rs149979685 variants.
Material and Methods
Sample Selection
Eleven AA subjects included in our previous study (Logue et al. 2014) who had at least one of the AKAP9 mutations for rs144662445 and rs149979685 (AKAP9+) and available lymphoblastoid cell lines were identified in the MIRAGE Study cell repository at Boston University and at the National Cell Repository for Alzheimer Disease (NCRAD) at Indiana University. All but one of these subjects had the mutation at both loci, while one subject had only the rs144662445 mutation. A comparison group of 17 AA subjects lacking the AKAP9 mutations (AKAP9−) with corresponding cell lines was identified from the same sources. An attempt was made to match the two groups for age, sex, AD status, and APOE genotype (Table 1). AKAP9+ and AKAP9− subjects had similar proportions of females (91.1% vs. 94.1%) and mean ages (76.8 vs. 78.5 years). The AKAP9+ group had a higher proportion of AD subjects (63.6% vs. 47.1%) but fewer APOE ε4 carriers (45.5% vs. 58.8%).
Construction of APPsw Lentiviral Plasmid and Lentivirus Stock Production
Full length human APP751 Swedish (APPsw) KM670/671NL mutant was PCR amplified (ClonAmp HiFi, Clontech) from APPsw in pcDNA3 vector (So et al. 2013) using the following primers: forward primer (5′ –TTACCTTCGAACCGCGGCTAGTTCTGCATCTGCTCAAAGAAC) and reverse primer (5′ – TAGCTCGAGTGCGGCCGCAACCCAG). Amplified product was subsequently cloned into the HFUW lentiviral vector (Chanoux et al. 2009) digested with EcoRI using In Fusion cloning kit (Clontech) following manufacturer’s protocol. The LV-APPsw plasmid was amplified in GCI-L3 E. coli cells (GeneCopoeia) to produce stable LV-APPsw plasmid. The plasmid was purified using ZymoPURE plasmid kit (Zymo). Lentivirus stocks were generated using the 293 T cell line co-transfected with LV-APPsw plasmid and two packaging plasmids (pMD2.G and psPAX2, Addgene #12259 and 12,260). Forty hours later, the supernatant was collected and filtered (0.45 mm) and stored frozen at −80 °C.
Lymphoblastoid Cell Lines (LCLs) and Virus-Mediated Gene Transduction
LCLs were maintained in RPMI 1640 (Invitrogen) with 10% FBS (Atlanta Biologicals) with 1× Penicillin-Streptomycin (Invitrogen). Cells (2 million cells in T25 flask) were infected by either 1:3 dilution of LV-APPsw lentiviral stock solution or 4 μL of recombinant replication-incompetent adenovirus type 5 expressing human 2N4R microtubule-associated protein Tau (1–441) for overnight at 37 °C, followed by replacing tissue culture medium as described (Sato et al. 2006).
Human Tau and pTau ELISA
After adenovirus infection, cells were subjected to 16 h incubation with 10 μM rolipram (CAS# 61413–54-5, Tocris, Minneapolis, MN), the PDE4 inhibitor that increases the intracellular cAMP level and activates AKAP-tethered PKA (McCahill et al. 2005), or control vehicle (0.5% dimethylsulfoxide in tissue culture medium). The cells were pelleted by centrifugation at 150 x g for 5 min at 4 °C, washed in ice-cold phosphate-buffered saline (PBS) and centrifuged, and the cell pellet was lysed in 5 volume of TENT buffer (20 mM Tris-Cl, pH 7.5, 2 mM ethylenediaminetetraacetic acid, 150 mM NaCl, 1% Triton X-100, 0.1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, and 5 mM Na3VO4, all from Sigma-Aldrich, St. Louis, MO). The cell lysates were placed on ice and vortexed for 20 s every 5 min for 20 min, followed by centrifugation at 22,000 x g for 30 min to remove insoluble materials. The supernatant was transferred to a new tube and subjected to the quantification of total proteins by BCA assay (Cat# PI-23221, Pierce/Thermofisher Scientific, Inc.), total human Tau by ELISA (human Tau ELISA kit, Cat# KHB0041, Life Technologies), and pT181 pTau by ELISA (pTau [pT181] Human ELISA Kit, Cat# KHO0631, Life Technologies) as described (Asai et al. 2015). The pT181 pTau values were normalized by the total Tau value for each sample.
APP and Aβ ELISA
After APPsw lentivirus infection for 48 h, the supernatants were collected for Aβ40 ELISA, and the cells were pelleted by centrifugation at 150 x g for 5 min at 4 °C, washed in ice-cold phosphate-buffered saline (PBS) and centrifuged, and the cell pellet was lysed in lysis buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 50 mM NaF, 1% Triton X-100 and 10 mM EDTA) with protease cocktail inhibitor tablet (1 tablet per 50 mL, Roche). The conditioned medium and cell lysates were analyzed by SDS-PAGE and western blotting using APP antibody (6E10 mAb) in the cell lysate for total APP as described previously (Jun et al. 2014). Primary antibody for western blots was mouse monoclonal 6E10 (Covance, 1:1000) against amino acids 1–17 of Aβ that also recognizes human APP. ELISA was carried out using the human Aβ40 ELISA kit (Invitrogen) in accordance with manufacturer’s protocol with samples diluted 1:2 in diluent buffer.
Statistical Analysis
The R statistical computing package (R Development Core Team 2008) was used to evaluate the association of AKAP9 genotype with measures of Aβ and phosphorylated Tau. Specifically, the influence of AKAP9 mutations (predictor) with normalized Aβ (Aβ/APP) and phosphorylated Tau (pTau/Tau) measures (outcomes) was analyzed in random-effects linear models including covariates for AD status and APOE genotype and a random effect term for sample to adjust for non-independence of repeated measures. The lmer function of lme4 R package (Bates et al. 2015) was used to fit the mixed models. Pairwise contrasts of differences between groups (e.g. AKAP9+, AD+ vs AKAP9+, AD−) were performed within the random-effects model framework using the multcomp R package (Hothorn et al. 2008) and results were illustrated using boxplots.
Human Tau IP and Mass-Spectrometry
After adenovirus infection, a subset of cell lines were subjected to 16 h incubation with 20 μM rolipram (CAS# 61413–54-5, Tocris, Minneapolis, MN). Cells were then pelleted by centrifugation at 300 x g for 5 min at 4 °C. Supernatant was then removed and cells were lysed in 300 μL of TENT++ buffer (50 mM Tris-HCl pH 8, 2 mM ethylenediaminetetraacetic acid (EDTA), 150 nM NaCl, 1% Triton X-100, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM NaF, and 1 mM Na3VO4. All chemicals were from Sigma, except Na3VO4, which was purchased from Alfa Aesar). Cells were vortexed for 1 min and then incubated on ice for 10 min, followed by centrifugation at 20,000 x g for 30 min at 4 °C to remove cell debris. The supernatant was then transferred to a fresh tube. BCA was used to quantify total protein amount. One mg of total protein was then processed for IP according to the instructions of the Pierce Direct IP column Kit with minor modifications (Cat #26148 Pierce/Thermofisher Scientific, Inc.). Columns used contained 10 μg of directly conjugated Tau13 antibody (provided by Binder/Kanaan laboratories) (Combs et al. 2016). Lysis buffer was replaced with TENT++. Samples were incubated in 45 μL NuPage LDS sample buffer (Cat# NP0007, Thermofisher Scientific, Inc.) for 20 min at 95 °C, and then eluted from the columns. The samples were then run for 1.5 cm in a 10% NuPage gel, and fixed for 30 min in 50% methanol, 40% miliQ water, and 10% acetic acid. The gels were then stained for 2 h in 50% methanol, 40% miliQ water, and 10% acetic acid and 0.25% w/v Coomassie brilliant blue R-250. Gels were destained overnight in 10% methanol, 7.5% acetic acid and 82.5% water. Following destaining, gels were washed twice for 10 min in miliQ water. Individual lanes were then extracted from the gel, stored in 10% acetic acid and sent to the mass spectrometry core at the University of Massachusetts Medical Center (Shrewsbury, MA) for processing.
Proteomic Analysis
Resulting proteomic data were analyzed using the Scaffold 4 software. The 441-amino acid isoform of Tau was analyzed for the presence of post translational modifications (PTMs), which were visualized as proportion of modified reads out of total reads. These data were then analyzed for differences in PTMs of Tau protein according to AKAP9 mutation or AD status using a paired t-test implemented in the Prism 6 software from Graphpad. The results of the t-tests were visualized in a paired differences graph where each point represents the proportion of modified reads in AKAP9AKAP9+ cells minus the proportion for the same site in AKAP9− cells or with AD+ lines minus the proportion for the same site in AD− cells. To understand functional differences in proteins co-precipitated with Tau, AKAP9+ or AKAP9− cell line proteins were filtered to retain only those proteins with at least two peptide spectra in all but one sample. Next, those protein expression levels were collapsed into a single number with the highest expression level being retained. Finally, the 400 most abundant proteins were selected in each condition and compared. Total abundance was determined by calculating normalized total spectra values using the iBAQ method within the Scaffold 4 software. The resulting 800 proteins were compared, and 51 emerged as unique to the top 400 of each condition (102 total). The 51 proteins from each condition were then analyzed by DAVID (Huang da et al. 2009a, b) to determine their ontologies. Only those ontologies with a p-value and False Discovery Rate (FDR, or q-value) less than 0.05 were retained. FDR was calculated by DAVID using the Benjamini method (Huang da et al. 2009a, b). Ontologies included are UP_KEYWORDS (UniProt Keywords), GOTERM_BP, CC, or MF_DIRECT (Gene ontology terms for Biological Processes, Cellular Component, or Molecular Function), and KEGG_Pathways (Kyoto Encyclopedia of Genes and Genomes).
Results
AKAP9 Mutations Do Not Affect APP Processing or Aβ Production
Our initial assessment of LCLs showed barely detectable secretion of endogenous human Aβ40 from these cell lines. Thus, we transduced recombinant human APP751 mutant carrying the KM670/671NL Swedish mutation into our LCLs. We examined the effect of AKAP9 (rs144662445 and rs149979685) mutations on the production of APP and Aβ40 in LCLs. Figure 1a shows the results of western blots (WB) of 28 LCLs transfected transiently with the APP mutant. The Aβ40 released to the medium was analyzed by ELISA, and normalized to the total APP intensity as determined by densitometry from the WB in Fig. 1a. A boxplot of the Aβ40/APP ratio by AKAP9 mutation and AD status is presented in Fig. 1b. The Aβ40/APP ratio was not significantly different as a function of AD status or AKAP9 or APOE genotype (p > 0.10; Table 2).

Measurement of Aβ40 production in lymphocytes infected with LV-APPsw. a Western Blot analysis of total APP in lentivirus-APPsw infected and control non-infected lymphocytes. A monoclonal antibody against APP (6E10) was used to detect endogenous and over expressed APP. The four groups tested were healthy controls (HC) APAK9−, HC APAK9+, AD APAK9−, AD APAK9+. APP transfected HEK cell lysate was used as a positive control for APP expression (+). b Mean Aβ40/APP ratio as a function of AD status and the presence of AD risk alleles for AKAP9 variants rs144662445 and rs149979685. An Aβ40/APP ratio was used since each cell line was transfected separately, thus the levels of Aβ40 were relative to the levels of transfected APP
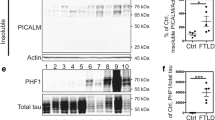
AKAP9 Mutations Increase Tau Phosphorylation in Rolipram-Treated Cells
We examined the effect of AKAP9 mutations on Tau phosphorylation in our cell lines. AKAP9 tethers PKA and regulates its sensitivity to cAMP for this activation (Terrin et al. 2012). Since the basal cAMP was insufficient to monitor the effect of PKA activation, the cells were treated with roliplram. Rolipram is a PDE4 inhibitor that increases the intracellular cAMP level and activates AKAP-tethered PKA, and has previously been shown to increase Tau phosphorylation in conjunction with PKA (McCahill et al. 2005). Figure 2 presents a boxplot of the pTau/Tau ratio as a function of AKAP9 and AD status for both the rolipram-treated and untreated samples. In the rolipram-untreated samples, none of the predictors are significant (all p > 0.09, Table 3a). In the mixed model of pTau/Tau for the treated samples (Table 3b), AKAP9 mutation carrier status was the only significant predictor, with AKAP9+ subjects having higher pTau ratio than AKAP9− subjects (β = 0.21, p = 2.50 × 10−6). Post-hoc pairwise group comparisons indicate that pTau/Tau was significantly higher in AKAP9+ subjects within both AD cases (β = 0.22, p = 2.61 × 10−6) and cognitively normal controls (β = 0.19, p = 0.00065).

Mean pTau/Tau ratio as a function of AD status and the presence of AD risk alleles for AKAP9 variants rs144662445 and rs149979685 in rolipram treated and untreated lymphoblastoid cell lines
Tau IP-Proteomics
We then wanted to further investigate differences in post-translational modicfications of Tau and how this might alter the makeup of Tau-interacting proteins. Table 4 shows the proportion of observed post-translational modifications (PTMs) out of total reads of the protein Tau at individual amino acids as a function of AD and AKAP9 mutation status following rolipram treatment for a subset of cell lines (n = 10). Differences in PTMs were then examined between AKAP9 mutation carriers and non-carriers (Fig. 3). Although no PTMs were significant after adjusting for multiple testing, several PTMs, such as pT69, pT212, and pS214, were nominally significant at p < 0.05 (Fig. 3 and Table 4). There were no differences in Tau phosphorylation PTMs between cell lines derived from AD cases and controls (Fig. S1a).

Proportions of post-translational modifications (PTMs) for reads of Tau protein derived from lymphocytes treated with rolipram as a function of AKAP9 mutation status. Plot shows proportion of reads with PTMs out of total as mean ± SEM of PTMs at each amino acid in the full-length 441 amino acid isoform of Tau. Residues with nominal p-values (less than 0.05) by t-test are indicated by a “^”
To help resolve patterns of Tau-interacting protein expression among AKAP9 + and − cells, we selected the top 400 enriched proteins that had at least two unique peptide spectra in all but one sample by Tau-mediated immunoprecipitation. We observed that 51 of these proteins were expressed only in AKAP9+ cells and other 51 proteins were expressed only in AKAP9− cells (Fig. 4, Tables S1 and S2). Analysis of the function of these proteins by DAVID revealed that although AKAP9 + and − cells contained distinct proteins, the proteins common to cells with and without AKAP9 mutations were enriched for acetylation, extracellular exosomes, poly(A) RNA binding, cytoplasm and phosphoproteins (Table S3). AKAP9− cell lines included proteins involved in RNA binding, methylation, the spliceosome, the nucleus, and protein binding (Table S4). In contrast, AKAP9− cell lines included proteins involved in the proteasome, ubiquitination, prenylation, and cilia formation, as well as in NF-kappaB, Wnt, T-cell receptors, and TNF and Fc epsilon signaling pathways (Table S5). Interestingly, only the AKAP9− cell lines showed enrichment for ontologies related to the mitotic cell cycle, echoing earlier findings that the AKAP9 mutations interfere with cell cycle progression (Logue et al. 2014).

Venn diagram of top Tau-interacting 400 proteins for AKAP9+ and AKAP9− cell lines identified by Tau immunoprecipitation. All included proteins had at least two unique peptide spectra in all but one sample, and then were collapsed into single protein identities taking the highest expression value. The top three proteins for each category (AKAP9+ unique, left; AKAP9+and AKA P9− common, center; AKAP9− unique, right). The top three proteins for AKAP9+ cell lines are involved in RNA binding (ERH, Enhancer of Rudimentary Homolog; SNRPD1, Small Nuclear Ribonucleotide Protein D1 polypeptide; RPL11, Ribosomal Protein L11). MAPT (the gene encoding Tau) is common to the top three proteins of both AKAP9+ and AKAP9− cell lines (as well as IGKV2D-28, Immunoglobulin Kappa Variable 2D-28 and RPL39, Ribosomal Protein L39). The top three proteins unique to AKAP9− cell lines include a member of the proteasome family, PSMD8 (Proteasomal 26S Subunit, Non-ATPase 8). Other top unique proteins include IGKC, Immunoglobulin Kappa Constant and HLA-DQA2, Major Histocompatibility Complex, Class II DQ Beta 2
Further ontological analysis revealed that acetylation, protein methylation, extracellular exosomes, and poly(A) RNA binding are common ontologies of Tau-interacting molecules in cell lines from both AD subjects and controls (Fig. S1b and Tables S6-S7). The most notable ontologies include phosphoproteins, mitochondria, and proteasomal molecules for proteins enriched only in the AD+ cell lines (Table S9). MHC class II receptors and other diseases such as lupus and tuberculosis were significant ontologies for proteins enriched only in cell lines from control subjects (Table S10).
Together, these findings suggest that the Tau-interactome can provide new insights about the effect of the AKAP9 rs144662445 and rs149979685 mutations on AD risk and the biological functions of Tau.
Discussion
This is one of only a few published functional genomic studies to assess the potential pathogenic effect of AD-associated genetic variants using primary lymphoblastoid cell lines (Johnston et al. 1997; Zou et al. 2013). To our knowledge, this is the first to study effects in cells from permutations of AD cases and cognitively normal controls with and without such variants. Our study leveraged phenotypic data previously assembled by the Alzheimer Disease Genetics Consortium for large-scale genetic studies in AAs (Reitz, et al. 2013) and genotypes for two AA-specific AKAP9 mutations (I2558M and S3771 L) in the ADGC cohort (Logue et al. 2014). Although multiple reports demonstrated the presence of AKAP9 in human T cells (Herter et al. 2015; Robles-Valero et al. 2010; Skalhegg et al. 2005), we did not detect endogenous Tau and the APP level was barely detectable in the LCLs. We compensated for the low expression of these two AD-related proteins by employing recombinant virus-mediated expression of human full-length Tau or the familial AD-linked APP Swedish mutant. We observed generation of Aβ after expression of APPsw, demonstrating the existence of β/γ processing machinery of APP in human LCLs.
Our data show that these two amino acid substitutions in AKAP9 had no effect on the amyloidogenic APP processing pathway that generates Aβ. This finding also suggests that the AKAP9 mutation(s) do not affect the interaction of AKAP9 with the regulatory subunit of PKA, nor do they alter the sensitivity of AKAP9 to cAMP for its activation.
The I2558M and S3771 L substitutions, however, significantly enhanced pTau levels in the presence of rolipram, the PDE4 inhibitor that increases the intracellular cAMP level. The increase in cAMP level triggered by rolipram mimics the NMDA-R dependent activation of excitatory pathways caused by cAMP-induced PKA activation seen early in the course of Alzheimer’s disease (van der Harg et al. 2017). Use of this drug allowed us to more readily understand the effect of the AKAP9 mutations on Tau phosphorylation. The effect on phosphorylation was independent of clinically manifest AD, sex, or APOE genotype in this AA cohort. Our finding suggests that one or both of these mutations have a direct effect on the intracellular signaling leading to phosphorylation of Tau. Specifically, the T69, T212, and S214 residues show a nominally significant increase in phosphorylation in AKAP9+ cell lines when treated with rolipram. S214 is reported to be a direct phosphorylation site of PKA (Carlyle et al. 2014). Overall, the total phosphorylation of Tau is increased in AKAP9+ cell lines as compared to the AKAP9− cell lines. Since this biological phenotype is dependent on rolipram treatment, we assume that these AKAP9 variants alter the sensitivity of PKA to cAMP, which results in the difference in the phosphorylation status of Tau. While many studies have shown beneficial effects of rolipram in AD model animals, high doses impaired prefrontal cortex function in aged monkeys, possibly owing to the different neurochemical needs of the prefrontal cortex in comparison with the hippocampus, and indicating that rolipram treatment may only benefit hippocampal functioning (Ramos et al. 2003, reviewed in Heckman et al. 2017). Furthermore, based on our results, rolipram treatment may not be beneficial in the long term, as we have found it to increase Tau phosphorylation.
Our findings suggest that altered AKAP9 can lead to an increase in Tau phosphorylation, which is associated with AD. The mutations we studied, located in exons 36 and 51, respectively, are rare, and thus do not contribute substantially to AD risk in the general population. Moreover, because rs144662445 and rs149979685 AKAP9 variants are co-segregating in nearly all subjects in this cohort, it is difficult to determine whether a particular mutation or the combination of both (I2558M or S3771 L) is responsible for the functional alteration. Only one AKAP9+ cell line had a single variant (I2558M). Although the treated pTau level in this cell line was in the range of scores observed in the other AKAP9+ cell lines, this is not enough information to perform a reliable statistical test. Vardarajan et al. recently observed association of AD risk with a distinct rare AKAP9 missense mutation in exon 8 (R434W) in Caribbean Hispanic families with multiple members affected by AD (unpublished results). Recently, a genome-wide association study of AD-related endophenotypes in the ADNI study cohort subjects of European ancestry with mild cognitive impairment revealed significant association of several common SNPs (minor allele frequency, MAF = 0.45 and located 381 kb upstream of AKAP9) with pTau level (p < 6.9 × 10−7 for all SNPs) (Chung et al. 2018). In the same study, there was strong evidence for association of rs149454736 (MAF = 0.03), which is located between exons 45 and 46 of AKAP9 and is only 1513 bp from rs149979865, with a brain MRI measure of hippocampal volume in AD subjects (p = 2.2 × 10−7). Further experiments are needed to determine whether these variants are functional and have an impact on Tau phosphorylation similar to the ones examined in this study.
Oxidation of the methionine residues of Tau was increased in rolipram-treated cells containing the AKAP9 mutations (Fig. 3 and Table 4, residues M11, 31, 250 and 419; M11 and 250 nominally significant). A previous study reported that Tau aggregation is promoted by oxidation of the C322 residue present in the R3 region of the repeating microtubule binding domains found in all Tau isoforms (Martin et al. 2011; Schweers et al. 1995). However, the novel methionine PTMs identified in this study are located outside of the R3 region and therefore would not contribute to aggregation in the same manner as C322.
Protein ontology analysis revealed that there are key differences in the Tau interactome between the AKAP9+ and AKAP9− cell lines. The AKAP9+ cells were enriched in Tau-interacting proteins involved in protein methylation and the methylosome. Methylation of Tau protein has been reported in hAPP transgenic mice at locations corresponding to homologous residues of the human Tau 441 isoform (R126, R155, K163, K259, K311, K331, and R349) (Morris et al. 2015). Interestingly, the AKAP9+ cell lines were enriched for RNA binding proteins, including riboproteins and spliceosome proteins. Such proteins have been implicated in the formation of stress granules which accumulate in AD and co-localize with sites of Tau pathology (Vanderweyde et al. 2016). The AKAP9− cell lines are enriched for ontologies involved with Rab small GTP-binding proteins, proteasomal complex and cell cycle progression, a function previously ascribed to AKAP9 (Logue et al. 2014). These findings suggest that the AKAP9 mutations may alter the Tau interactome and, consequently, alter its metabolism from Rab-mediated endosomal trafficking and proteasomal degradation and onto RNA binding protein-mediated aggregation in the cells.
There are limitations to our study worth noting. Although our previous study ruled out the possibility of other coding mutations in AKAP9 or adjacent genes that are in linkage disequilibrium with the rs144662445 and rs149979685 as the functional AD-related variant at this locus (Logue et al. 2014), the true causal variant may be in a non-coding regulatory region. This idea is consistent with our recent observation that a SNP in the KANSL1 gene promoter accounts for the AD association peak with MAPT (the gene encoding Tau) and co-regulates expression of alternatively spliced exon 3 in MAPT (Jun et al. 2016). Another potential concern about these results is that AKAP9 expression in lymphoblastoid cell lines may not hold true in neurons. Although further experiments in neuronal cells in culture or from brain tissue is a logical next step, the naturally occurring mutations studied here are rare (and thus unlikely to be found in brain repositories) and introducing them into cultured cells which lack important features of the genetic background may not mimic in vivo conditions. It was also somewhat surprising that we did not detect a difference in endogenous expression of APP in LCLs from AD cases compared to controls. This observation is consistent with a previous study which also did not find a difference in APP expression between non-familial AD cases and controls (Johnston et al. 1997). In addition, we did not observe endogenous expression of Tau in LCLs which, to our knowledge, has not been reported. It is possible that the lack of expression differences or expression per se of these proteins is due to their variable roles in LCLs and neurons. Alternatively, differential expression of Tau and APP in LCLs may be detectable only when adjusting for genetic influences (e.g., AKAP9 mutations) that directly impact their expression.
In conclusion, this study showed that lymphoblastoid cell lines derived from persons possessing the AKAP9 I2558M and/or S3771 L variants exhibit significant alteration in phosphorylation of recombinant Tau protein after virus-mediated transient expression of Tau and chemical inhibition of PDE4 by rolipram. This alteration in Tau phosphorylation was independent of AD status and APOE genotype. The presence of AKAP9 mutations altered the composition of Tau-interacting proteins, increasing interaction of RNA and protein binding molecules, while proteasomal proteins were enriched in cell lines lacking these mutations. The approach of using human-derived cell lines for ex-vivo evaluation of pathogenic effects of AD-associated polymorphisms could be widely applicable to other diseases as a new modality of functional genomics, in addition to 3-dimensional tissue culture models of human neuronal cells (Choi et al. 2014; Kim et al. 2015; Shi et al. 2012).
References
Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K, Hermjakob H (2010) The IntAct molecular interaction database in 2010. Nucleic Acids Res 38(Database issue):D525–D531
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593
Bates D, Machler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Software 67:1–48
Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Papetti A, Stuani C, Di Luca M, Gennarelli M, Padovani A (2009) Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat 30:E974–E983
Carlyle BC, Nairn AC, Wang M, Yang Y, Jin LE, Simen AA, Ramos BP, Bordner KA, Craft GE, Davies P, Pletikos M, Sestan N, Arnsten AF, Paspalas CD (2014) cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A 111:5036–5041
Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ (2009) ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem 284:5994–6003
Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS (2007) Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A 104:20990–20995
Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY (2014) A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515:274–278
Chung J, Wang X, Maruyama T, Ma Y, Zhang X, Mez J, Sherva R, Takeyama H, (2018) The alzheimer’s disease neuroimaging initiative, In: Lunetta KL, Farrer LA, Jun GR (eds) Genome-wide association study of Alzheimer disease endophenotypes at prediagnosis stages. Alzheimers Dement. https://doi.org/10.1016/j.jalz.2017.11.006
Combs B, Hamel C, Kanaan NM (2016) Pathological conformations involving the amino terminus of tau occur early in Alzheimer's disease and are differentially detected by monoclonal antibodies. Neurobiol Dis 94:18–31
Hadar A, Milanesi E, Squassina A, Niola P, Chillotti C, Pasmanik-Chor M, Yaron O, Martásek P, Rehavi M, Weissglas-Volkov D, Shomron N, Gozes I, Gurwitz D (2016) RGS2 expression predicts amyloid-β sensitivity, MCI and Alzheimer's disease: genome-wide transcriptomic profiling and bioinformatics data mining. Transl Psychiatry 6(10):e909
Heckman PRA, Blokland A, Prickaerts J (2017) From age-related cognitive decline to Alzheimer’s disease: a translational overview of the potential role for phosphodiesterases. Adv Neurobiol 17:135–168
Herter JM, Grabie N, Cullere X, Azcutia V, Rosetti F, Bennett P, Herter-Sprie GS, Elyaman W, Luscinskas FW, Lichtman AH, Mayadas TN (2015) AKAP9 regulates activation-induced retention of T lymphocytes at sites of inflammation. Nat Commun 6:10182
Hothorn T, Bretz F, Westfall P (2008) Simultaneous inference in general parametric models. Biom J 50:346–263
Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J (2006) Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics 7:118
Huang da W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Huang da W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Johnston JA, Lannfelt L, Wiehager B, O'Neill C, Cowburn RF (1997) Amyloid precursor protein heat shock response in lymphoblastoid cell lines bearing presenilin-1 mutations. Biochim Biophys Acta 1362:183–192
Jun G, Asai H, Zeldich E, Drapeau E, Chen C, Chung J, Park JH, Kim S, Haroutunian V, Foroud T, Kuwano R, Haines JL, Pericak-Vance MA, Schellenberg GD, Lunetta KL, Kim JW, Buxbaum JD, Mayeux R, Ikezu T, Abraham CR, Farrer LA (2014) PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann Neurol 76:379–392
Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert J-C, Chung J, Naj AC, Kunkle BW, Wang L-S, Bis JC, Bellenguez C et al (2016) A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21:108–117
Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I, Tsujita T, Okazaki Y, Nanko S, Kunugi H, Sasaki T, Kato T (2003) Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet 35:171175
Kim YH, Choi SH, D'Avanzo C, Hebisch M, Sliwinski C, Bylykbashi E, Washicosky KJ, Klee JB, Brustle O, Tanzi RE, Kim DY (2015) A 3D human neural cell culture system for modeling Alzheimer's disease. Nat Protoc 10:985–1006
Liu F, Liang Z, Shi J, Yin D, El-Akkad E, Grundke-Iqbal I, Iqbal K, Gong CX (2006) PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett 580:6269–6274
Logue MW, Schu M, Vardarajan BN, Farrell J, Bennett DA, Buxbaum JD, Byrd GS, Ertekin-Taner N, Evans D, Foroud T, Goate A, Graff-Radford NR, Kamboh MI, Kukull WA, Manly JJ, Alzheimer Disease Genetics Consortium, Haines JL, Mayeux R, Pericak-Vance MA, Schellenberg GD, Lunetta KL, Baldwin CT, Fallin MD, Farrer LA (2014) Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimers Dement 10:609–618
Luna-Munoz J, Chavez-Macias L, Garcia-Sierra F, Mena R (2007) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer's disease. J Alzheimers Dis 12:365–375
Martin L, Latypova X, Terro F (2011) Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int 58:458–471
McCahill A, McSorley T, Huston E, Hill EV, Lynch MJ, Gall I, Keryer G, Lygren B, Tasken K, van Heeke G, Houslay MD (2005) In resting COS1 cells a dominant negative approach shows that specific, anchored PDE4 cAMP phosphodiesterase isoforms gate the activation, by basal cyclic AMP production, of AKAP-tethered protein kinase A type II located in the centrosomal region. Cell Signal 17:1158–1173
Middleton FA, Pato CN, Gentile KL, McGann L, Brown AM, Trauzzi M, Diab H, Morley CP, Medeiros H, Macedo A, Azevedo MH, Pato MT (2005) Gene expression analysis of peripheral blood leukocytes from discordant sib-pairs with schizophrenia and bipolar disorder reveals points of convergence between genetic and functional genomic approaches. Am J Med Genet B Neuropsychiatr Genet 136:1225
Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, Mucke L (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 18:1183–1189
R Development Core Team (2008) R Core Team. R A language and environment for statistical computing 2014. R Foundation for Statistical Computing, Vienna
Ramos BP, Birnbaum SG, Lindenmayer I, Newton SS, Duman RS, Arnsten AF (2003) Dysregulation of protein kinase a signaling in the aged prefrontal cortex: new strategy for treating age-related cognitive decline. Neuron 40:835–845
Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, Valladares O, Lin CF, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, Buxbaum JD, Murrell JR, Raj T, Ertekin-Taner N, Logue M, Baldwin CT, Green RC, Barnes LL, Cantwell LB, Fallin MD, Go RC, Griffith P, Obisesan TO, Manly JJ, Lunetta KL, Kamboh MI, Lopez OL, Bennett DA, Hendrie H, Hall KS, Goate AM, Byrd GS, Kukull WA, Foroud TM, Haines JL, Farrer LA, Pericak-Vance MA, Schellenberg GD, Mayeux R, Alzheimer Disease Genetics Consortium (2013) Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 309:1483–1492
Robles-Valero J, Martin-Cofreces NB, Lamana A, Macdonald S, Volkov Y, Sanchez-Madrid F (2010) Integrin and CD3/TCR activation are regulated by the scaffold protein AKAP450. Blood 115:4174–4184
Sato S, Cerny RL, Buescher JL, Ikezu T (2006) Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J Neurochem 98:1573–1584
Schweers O, Mandelkow EM, Biernat J, Mandelkow E (1995) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci U S A 92:8463–8467
Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ (2012) A human stem cell model of early Alzheimer's disease pathology in down syndrome. Sci Transl Med 4:124
Skalhegg BS, Funderud A, Henanger HH, Hafte TT, Larsen AC, Kvissel AK, Eikvar S, Orstavik S (2005) Protein kinase A (PKA)--a potential target for therapeutic intervention of dysfunctional immune cells. Curr Drug Targets 6:655–664
So PP, Khodr CE, Chen CD, Abraham CR (2013) Comparable dimerization found in wildtype and familial Alzheimer's disease amyloid precursor protein mutants. Am J Neurodegener Dis 2:15–28
Terrin A, Monterisi S, Stangherlin A, Zoccarato A, Koschinski A, Surdo NC, Mongillo M, Sawa A, Jordanides NE, Mountford JC, Zaccolo M (2012) PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J Cell Biol 198:607–621
The 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Tsuang MT, Nossova N, Yager T, Tsuang MM, Guo SC, Shyu KG, Glatt SJ, Liew CC (2005) Assessing the validity of blood-based gene expression profiles for the classification of schizophrenia and bipolar disorder: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 133:15
van der Harg JM, Eggels L, Bangel FN, Ruigrok SR, Zwart R, Hoozemans JJM, la Fleur SE, Scheper W (2017) Insulin deficiency results in reversible protein kinase a activation and tau phosphorylation. Neurobiol Dis 103:163–173
Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PE, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, Li H, Petrucelli L, Wolozin B (2016) Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep 15:1455–1466
Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R, Gstaiger M (2013) The protein interaction landscape of the human CMGC kinase group. Cell Rep 3:1306–1320
Vawter MP, Ferran E, Galke B, Cooper K, Bunney WE, Byerley W (2004) Microarray screening of lymphocyte gene expression differences in a multiplex schizophrenia pedigree. Schizophr Res 67:4152
Witczak O, Skalhegg BS, Keryer G, Bornens M, Tasken K, Jahnsen T, Orstavik S (1999) Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450. EMBO J 18:1858–1868
Wu G, Feng X, Stein L (2010) A human functional protein interaction network and its application to cancer data analysis. Genome Biol 11:R53
Zou F, Belbin O, Carrasquillo MM, Culley OJ, Hunter TA, Ma L, Bisceglio GD, Allen M, Dickson DW, Graff-Radford NR, Petersen RC, Genetic, Environmental Risk for Alzheimer's Disease Consortium, Morgan K, Younkin SG (2013) Linking protective GAB2 variants, increased cortical GAB2 expression and decreased Alzheimer's disease pathology. PLoS One 8(5):e64802
Acknowledgements
This study was funded in-part by grants from the National Institute on Aging (NIA, R01-AG048927, U01-AG032984, P30-AG13846, and RF1-AG054199) and Alzheimer’s Association (DVT-14320835 and AARF-16442664). Biological samples used in this study were prepared by the Molecular Genetics Core Facility at Boston University (Irene Simkin, Manager) or obtained from the National Cell Repository for Alzheimer’s Disease (NCRAD) at Indiana University funded by NIA (U24-AG021886). We would also like to thank Dr. John Leszyk at the Proteomics and Mass Spectrometry Facility, University of Massachusetts Medical School.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ikezu, T., Chen, C., DeLeo, A.M. et al. Tau Phosphorylation is Impacted by Rare AKAP9 Mutations Associated with Alzheimer Disease in African Americans. J Neuroimmune Pharmacol 13, 254–264 (2018). https://doi.org/10.1007/s11481-018-9781-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-018-9781-x