
Abstract
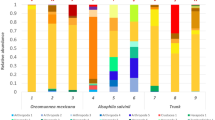
A number of basic and applied questions in ecology and environmental management require the characterization of soil and leaf litter faunal diversity. Recent advances in high-throughput sequencing of barcode-gene amplicons (‘metabarcoding’) have made it possible to survey biodiversity in a robust and efficient way. However, one obstacle to the widespread adoption of this technique is the need to choose amongst many candidates for bioinformatic processing of the raw sequencing data. We compare three candidate pipelines for the processing of 18S small subunit rDNA metabarcode data from solid substrates: (i) USEARCH/CROP, (ii) Denoiser/UCLUST, and (iii) OCTUPUS. The three pipelines produced reassuringly similar and highly correlated assessments of community composition that are dominated by taxa known to characterize the sampled environments. However, OCTUPUS appears to inflate phylogenetic diversity, because of higher sequence noise. We therefore recommend either the USEARCH/CROP or Denoiser/UCLUST pipelines, both of which can be run within the QIIME (Quantitative Insights Into Microbial Ecology) environment.
Article PDF
Similar content being viewed by others

Avoid common mistakes on your manuscript.
References
Edgar R C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 2010, 26: 2460–2461
Hao X, Jiang R, Chen T. Clustering 16S rRNA for OTU prediction: a method of unsupervised Bayesian clustering. Bioinformatics, 2011, 27: 611–618
Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods, 2010, 7: 668–669
Fonseca V G, Carvalho G R, Sung W, et al. Second-generation environmental sequencing unmasks marine metazoan biodiversity. Nat Commun, 2010, 1: 98
Pruesse E, Quast C, Knittel K, et al. 2007 SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res, 2007, 35: 7188–7196
Munch K, Boomsma W, Huelsenbeck J, et al. Statistical assignment of DNA sequences using Bayesian phylogenetics. Syst Biol, 2008, 57: 750–757
Caporaso J G, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods, 2010b, 7: 335–336
Hamilton H C, Strickland M S, Wickings K, et al. Surveying soil faunal communities using a direct molecular approach. Soil Biol Biochem, 2009, 41: 1311–1314
Wu T, Ayres E, Bardgett R D, et al. Molecular study of worldwide distribution and diversity of soil animals. Proc Natl Acad Sci USA, 2011, 108: 17720–17725
Bienert F, De Danieli S, Miquel C, et al. Tracking earthworm communities from soil DNA. Mol Ecol, 2012, 21: 2017–2030
Epp L S, Boessenkool S, Bellemain E P, et al. New environmental metabarcodes for analysing soil DNA: potential for studying past and present ecosystems. Mol Ecol, 2012, 21: 1821–1833
Porazinska D L, Giblin-Davis R M, Esquivel A. Ecometagenetics confirms high tropical rainforest nematode diversity. Mol Ecol, 2010a, 19: 5521–5530
Porazinska D L, Giblin-Davis R M, Faller L, et al. Evaluating high-throughput sequencing as a method for metagenomic analysis of nematode diversity. Mol Ecol Resources, 2009, 9: 1439–1450
Porazinska D L, Sung W, Giblin-Davis R M, et al. Reproducibility of read numbers in high-throughput sequencing analysis of nematode community composition and structure. Mol Ecol Resources, 2010b, 10: 666–676
Taberlet P, Coissac E, Hajibabaei M, et al. Environmental DNA. Mol Ecol, 2012, 21: 1789–1793
Yoccoz N G, Bråthen K A, Gielly L, et al. DNA from soil mirrors plant taxonomic and growth form diversity. Mol Ecol, 2012, 21: 3647–3655
Koskinen J P, Holm L. SANS: high-throughput retrieval of protein sequences allowing 50% mismatches. Bioinformatics, 2012, 28: 438–443
Bik H M, Porazinska D L, Creer S, et al. Sequencing our way towardsunderstanding global eukaryoticbiodiversity. Cell, 2012, 27: 4
Smith B C, McAndrew T, Chen Z, et al. The cervical microbiome over 7 years and a comparison of methodologies for its characterization. PloS one, 2012, 7: 7
Lenz T, Becker S. Simple approach to reduce PCR artefact formation leads to reliable genotyping of MHC and other highly polymorphic loci—implications for evolutionary analysis. Gene, 2008, 427: 117–123
Coissac E, Riaz T, Puillandre N. Bioinformatic challenges for DNA metabarcoding of plants and animals. Mol Ecol, 2012, 21: 1834–1847
Taberlet P, Prud’Homme S M, Campione E, et al. Soil sampling and isolation of extracellular DNA from large amount of starting material suitable for metabarcoding studies. Mol Ecol, 2012, 21: 1816–1820
Yoccoz N G, Bråthen K A, Gielly L, et al. DNA from soil mirrors plant taxonomic and growth form diversity. Mol Ecol, 2012, 21: 3647–3655
Creer S, Fonseca V G, Porazinska D L, et al. Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Mol Ecol, 2010, 19: 4–20
Somerfield P J, Warwick R M, Moens T. Meiofauna techniques. In: Methods for the Study of Marine Benthos. Oxford: Blackwell Science Ltd., 2005. 229–272
Folmer O, Black M, Hoeh W, et al. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Marine Biol Biotechnol, 1994, 3: 294–299
Edgar R C, Haas B J, Clemente J C, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 2011, 27: 2194–2200
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 2006, 22: 1658–1659
Yu D W, Ji Y Q, Emerson B C, et al. Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol Evol, 2012, 3: 613–623
Haas B J, Gevers D, Earl A M, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res, 2011, 21: 494–504
Chou H H, Holmes M H. DNA sequence quality trimming and vector removal. Bioinformatics, 2001, 17: 1093–1104
Edgar R C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res, 2004, 32: 1792–1797
Amend A S, Seifert K A, Bruns T D. Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol Ecol, 2010, 19: 5555–5565
Gotelli N J, Colwell R K. Estimating species richness. In: Meagurran A E, McGill B J, eds. Biological Diversity: Frontiers in Measurement and Assessment. Oxford: Oxford University Press, 2011. 39–54
Nipperess D. Phylocurve: an R function for generating a rarefaction curve of phylogenetic diversity. http://davidnipperess.blogspot.com/2012/07/phylocurve-r-function-for-generating.html, 2011
R Development Core Team. R: A language and environment for statistical computing. In: R Foundation for Statistical Computing, Vienna, Austria, 2012
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol, 2003, 52: 696–704
Kearse M, Moir R, Wioson A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinf Appl Note, 2012, 28: 1647–1649
Yang X D, Sha L Q. Species composition and diversity of soil mesofauna in the ‘Holy Hills’ fragmentary tropical rain forest of Xishuangbanna, China. Chin J Appl Ecol, 2010, 12: 261–265
Caporaso J G, Bittinger K, Bushman F D, et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics, 2010a, 26: 266–267
Faith D P, Baker A M. Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol Bioinf Online, 2006, 2: 121–128
Matsen F A, Kodner R B, Armbrust E V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 2010, 11: 538
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published with open access at Springerlink.com
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Yang, C., Ji, Y., Wang, X. et al. Testing three pipelines for 18S rDNA-based metabarcoding of soil faunal diversity. Sci. China Life Sci. 56, 73–81 (2013). https://doi.org/10.1007/s11427-012-4423-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11427-012-4423-7