
Abstract
Purpose
In the present study we characterized a series of synthetic cannabinoids containing various heterocyclic scaffolds that had been identified as constituents of “Spice”, a preparation sold on the illicit drug market. All compounds were further investigated as potential ligands of the orphan receptors GPR18 and GPR55 that interact with some cannabinoids.
Methods
The compounds were studied in radioligand binding assays to determine their affinity for human cannabinoid CB1 and CB2 receptors expressed in CHO cells, and in cAMP accumulation assays to study their functionality.
Results
Structure-activity relationships were analyzed. The most potent CB1 receptor agonist of the present series MDMB-FUBINACA (12) (Ki = 98.5 pM) was docked into the human CB1 receptor structure, and a plausible binding mode was identified showing high similarity with that of the co-crystallized THC derivatives. MDMB-CHMCZCA (41) displayed a unique profile acting as a full agonist at the CB1 receptor subtype, but blocking the CB2 receptor completely. Only a few weakly potent antagonists of GPR18 and GPR55 were identified, and thus all compounds showed high CB receptor selectivity, mostly interacting with both subtypes, CB1 and CB2.
Conclusions
These results will be useful to assess the compounds’ toxicological risks and to guide legislation. Further studies on 41 are warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A challenging issue for forensic toxicologists and law makers is how to effectively respond to the constantly changing new psychoactive substances on the illicit drug market [1]. Among these, synthetic cannabinoids feature prominently [2, 3]. Between 2008, when so-called “Spice” products [4] containing synthetic cannabinoids began to appear on the drug market, and 2016, 169 new synthetic cannabinoids were confiscated and identified [2]. Most of them were discovered as powders, often in bulk amounts, while others were found in preparations of plant materials, e.g., minced herbs, onto which solutions of the cannabinoids had been sprayed [5]. These substances have been shown to bind to and in many cases activate cannabinoid (CB) receptors. CB receptors are divided into two subtypes, CB1 and CB2, which belong to the large family of rhodopsin-like class A G protein-coupled receptors (GPCRs) [6]. Both CB receptor subtypes are coupled to Gi proteins including a reduction in intracellular cAMP levels. The main psychoactive effects of cannabinoids are mediated by the CB1 receptor, which is predominantly expressed in the central nervous system [7], while CB2 receptor expression in the brain is restricted to microglial cells [8, 9]. CB2 receptors are highly expressed in the immune system, for example in tonsils and spleen [10, 11]. Activation of the CB2 receptor is considered as a new therapeutic option for the treatment of inflammatory diseases and pain [12, 13].
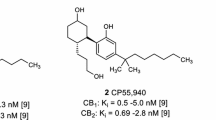
The plant-derived partial CB1 and CB2 receptor agonist Δ9-tetrahydrocannabinol (Δ9-THC, 1, Fig. 1) is used in therapy to target muscle spasms, nausea and cachexia [14]. The synthetic compound CP55,940 (2, Fig. 1) represents a potent full agonist at both receptor subtypes. A CB1 receptor antagonist, rimonabant, had been approved for the treatment of obesity but was later withdrawn from the market due to side effects resulting in depression and an increased suicide rate [15].

The prevalence for the use of illegal psychoactive substances in Europe by 15–16 year-old teenagers was estimated in 2015 to be about 4% [5]. Synthetic CB1 receptor agonists are abused as an alternative to natural marijuana due to their psychoactive and analgesic effects. For synthetic cannabinoids more and more severe side effects and intoxications are reported; they are predominantly neurologic symptoms, but acute organ toxicity has also been observed [16]. In the USA, the principle of enumeration is used to restrict newly discovered synthetic cannabinoids, and every single synthetic cannabinoid has to be individually listed by name in the US List of Schedule I drugs [17]. In Germany new synthetic cannabinoids are legally controlled since November 2016 when the “Neue-Psychoaktive-Stoffe-Gesetz” (NpSG, New Psychoactive Substances Act) was adopted in [18]. Similar regulations exist in Austria and Switzerland [19, 20]. All corresponding compounds, the chemical structures of which are represented by a general formula in the statute with known structure-activity relationships (SARs), were restricted. Newly discovered SARs of synthetic cannabinoids will, therefore, provide a basis for future amendments. However, in many cases, only limited information is available regarding the activity of new substances. Both the affinity of a drug for its receptor and its ability to produce an agonistic response are important features, and these should be determined according to a compound’s chemical structure. For important classes of synthetic cannabinoids, at least four structural components, which have firstly been described by Huffman et al. and were later refined by the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), are of importance (see Fig. 2 [3]): (1) a heterocyclic core consisting of indole or indazole with different substitutions; (2) a linker, e.g., an ester, amide or ketone; (3) a bulky lipophilic residue (R1), e.g., a heterocyclic or aryl substituent, but in newer synthetic cannabinoids a lipophilic amino acid can also be found; and (4) a residue (R2) which is a hydrophobic “side chain” attached to the nitrogen atom of the indole or the indazole ring system [21, 22]. The compound JWH-018 (3, see Fig. 1), a potent CB1 and CB2 receptor agonist, displays the basic features of this compound class and was one of the first synthetic cannabinoids identified in herbal blends for abuse [23, 24]. The common features of known synthetic cannabinoids are depicted in Fig. 2.

Common structural features of synthetic cannabinoids. The figure was adopted from the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) [3] and modified
In a previous study [25], we had determined the pharmacological properties of 48 synthetic cannabinoids collected by the Institute of Forensic Medicine of the University of Bonn. In the present study, we investigated the affinities and functional properties of a new series of 42 synthetic cannabinoids, 16 of which have not been reported as cannabinoid receptor ligands before. The investigated set of compounds comprises four different core structures. The first three groups (A, B, C, see Table 1) represent differently substituted indoles and indazoles, which are structurally derived from the synthetic cannabinoids previously introduced by Huffman et al. and are widely distributed in illicitly sold "Spice" products. In the current study we investigated compounds with l-valinamide (AB)/l-tert-leucinamide (ADB or MAB), methyl-3,3-dimethylbutanoate (MDMB), methyl-3-methylbutanoate (MMB), and 2-methyl-2-phenylpropyl (cumyl) moieties as substituents in the R1 position. Further classes of compounds consist of carbazoles (E), substituted in position 3, and benzimidazole derivatives (F).
Radioligand binding and cAMP functional studies on CB1 and CB2 receptors were complemented by CB1 receptor modeling and docking of the most potent CB1 receptor agonist of the present series to predict its interactions. We further tested all compounds for their ability to activate or block the two orphan GPCRs GPR18 and GPR55, both of which are known to interact with cannabinoids [26,27,28,29]. We discuss SARs of the newly investigated compounds, integrating previously reported data, thereby providing a comprehensive analysis, which will help to predict properties of novel derivatives.
Methods
Compounds
All compounds except for MDMB-CHMCZCA (41) were obtained from Cayman Chemical (Ann Arbor, MI, USA). According to the manufacturer, the purity of all compounds was declared to be > 95% as determined by liquid chromatography–tandem mass spectrometry (LC–MS/MS). MDMB-CHMCZCA (41) was purchased from www.brc-finechemicals.com. We confirmed the purity of all compounds in our laboratories by liquid chromatography–ultraviolet-mass spectrometry (LC–UV-MS) measurements and found it to be generally ≥ 96%, except for MDMB-FUBINACA (12, 93%) and Cl-2201 (37, 86%).
Radioligand binding assays
Radioligand binding assays were performed as described previously [25]. Membrane preparations of Chinese hamster ovary (CHO) cells overexpressing the human CB receptor subtype CB1 or CB2 were incubated in the presence of the test compound and the radioligand [3H]CP55,940 (0.1 nM, see Fig. 1) (Perkin-Elmer Life Sciences, Rodgau-Jügesheim, Germany), for 2 h. Bound and unbound radioligand were separated by rapid filtration through glass fiber GF/C-filters (Perkin-Elmer, Boston, MA, USA), using a Brandel 96-well Harvester (Brandel, Gaithersburg, MD, USA). Radioactivity on the filters was determined by liquid scintillation counting. Three separate experiments were performed, each in duplicates.
cAMP accumulation assays
cAMP accumulation assays were performed also as previously described [25]. Briefly, CHO cells stably expressing the respective human CB receptor subtype CB1 or CB2 were seeded overnight. Then the phosphodiesterase inhibitor Ro-20-1724 [4-(3-butoxy-4-methoxyphenyl)methyl-2-imidazolidone; Sigma-Aldrich, St. Louis, MO, USA], and subsequently the test compound (10–1 µM) and forskolin (10 µM, Sigma-Aldrich) were added. After incubation for 15 min the buffer was removed, and the cells were lyzed. The amount of cAMP was determined in a radioligand binding assay by incubating 50 µL of the cell lysate with 3 nM [3H]cAMP in the presence of protein preparations from bovine adrenal glands (cAMP binding protein) [30]. Bound and unbound radioligand were separated by rapid filtration through GF/B filters, and radioactivity was determined by liquid scintillation counting. To test for antagonistic activity, test compounds were added to Hank’s buffered salt solution (HBSS) containing 10% dimethyl sulfoxide (DMSO), 10 min after the application of Ro-20-1724, and the mixture was incubated for 20 min at 37 °C. Then, the CB agonist CP55,940 was added at a concentration of 0.03 µM, and cAMP determination was carried out as described above [25].
β-Arrestin assays
β-Arrestin assays were performed in recombinant CHO cells expressing either the human GPR18 or the human GPR55 as described before using the β-galactosidase enzyme fragment complementation technology (β-arrestin PathHunter™ assay; DiscoverX, Fremont, CA, USA) [25].
Data analysis
Data were analyzed using GraphPad Prism Version 4.02-6.1, (GraphPad Software, San Diego, CA, USA).
Molecular docking
Molecular docking studies were carried out with the software package Rosetta (www.RosettaCommons.org) using the 2017.08.59291 build [31, 32]. As templates the X-ray structures 5XRA and 5XR8 were employed [33]; fusion proteins and ligands were deleted and a conformer of MDMB-FUBINACA (12) was manually positioned in an initial model using the PyMOL Molecular Graphics System, Version 1.7.4.5 (Schrödinger, Inc., New York, NY, USA). A conformer library of MDMB-FUBINACA (12) was calculated using the BCL Conformer:Generator [34]. Docking procedure and scripts for data processing are described in supplementary material. Docking scores were calculated using the Rosetta InterfaceAnalyzer. The best scoring models were clustered into a set of plausible binding poses. Results were compared to the pose of THC-like agonists in the template crystal structures 5XRA and 5XR8 and displayed using UCSF Chimera [35].
Results and discussion
Cannabinoid CB1 and CB2 receptor affinities
In the present study, CB1 and CB2 receptor affinities of a new series of synthetic cannabinoids were determined in radioligand binding studies, which provide an ideal basis for the analysis of SARs (Table 1). The investigated compounds comprise indole, indazole, benzimidazole and carbazole derivatives. For some of the compounds, EC50 values had previously been determined by functional assays; however, functional data are highly dependent on the expression level of the receptors or “receptor reserve”, while Ki values obtained in binding studies are largely independent of the employed cellular background [36].
The present set of compounds includes amino acid derivatives. These types of compounds were originally described in a patent and claimed as potential pain therapeutics [37]. In all cases, an alkyl or heteroaryl residue was introduced as R2, and the amino acid was coupled to an amino group in the R1 position (see Table 1; Fig. 2) [37]. The presented compounds feature a pentyl or 5-fluoropentyl side chain in position R2 (for Table 1; Fig. 2). MMB-018 (5), an indole derivative substituted with a valine methyl ester, showed affinity in the low nanomolar range with a Ki value of 15.1 nM at the CB1 receptor and an almost identical Ki value of 14.0 nM at the CB2 receptor. The corresponding indazole AMB (6) was more potent displaying subnanomolar affinity for both CB receptor subtypes (CB1 Ki = 0.866 nM; CB2 Ki = 0.973 nM), indicating the superiority of the indazole core. The 5-fluoropentyl derivatives MMB-2201 (7) and 5F-AMB (8) were similarly as potent as their pentyl analogues MMB-018 (5) and AMB (6), respectively, showing that the terminal fluorination of the pentyl side chain gives almost no effect. Compounds with a p-fluorobenzyl residue or a bioisosteric cyclohexylmethyl residue showed increased affinities in the subnanomolar range in the indazole series (FUB-AMB (9), CB1 Ki = 0.387 nM, and MA-CHMINACA (10), CB1 Ki = 0.339 nM) and were about equipotent at the CB2 receptor. Banister et al. [38] had already investigated these compounds and also 5F-AMB (8) in a fluorescence-based membrane potential assay and determined potencies in the nanomolar range (EC50 values ranging from 1.9 to 71 nM) in that assay, while our radioligand binding assay revealed higher affinities.
The valine methyl ester was replaced by a tert-leucine methyl ester in four of the investigated compounds: 5F-ADB (11), MDMB-FUBINACA (12), MDMB-CHMICA (13) and MDMB-CHMINACA (14), substituted with each 5-fluoropentyl (11), p-fluorobenzyl (12) and cyclohexylmethyl residue (13,14) for R2, respectively. MDMB-FUBINACA (12) was the most potent compound of the entire set of investigated compounds with a Ki value of 0.0985 nM at the CB1 receptor and 0.130 nM at the CB2 receptor. Banister et al. [38] had reported EC50 values of 3.9 nM at CB1 and of 55 nM at CB2 receptors determined in a fluorescence-based membrane potential assay for this compound [38]. MDMB-FUBINACA had caused the highest hypothermal response which the authors had ever observed in rats [38]. These results showed once more that functional assays often do not correctly predict compounds’ affinities. MDMB-CHMICA (13), which also showed subnanomolar affinities for CB1 and CB2 receptors, was previously found to be involved in fatal intoxications, and it was concluded that the compound could cause multiple organ failure with lethal outcome in combination with alcohol [39, 40]. The corresponding indazole MDMB-CHMINACA (14) again showed even slightly higher affinities for both receptors.
Next, compounds with a valinamide substitution (R1) were studied. These were somewhat less potent than the valine methyl esters [compare 5F-AB-PICA (15)/MMB-2201 (7); AB-CHMINACA (20)/MA-CHMINACA (10); and 5F-AB-PINACA (16)/5F-AMB (8)]. 5F-AB-PICA (15), a 5F-pentyl-indole derivative, displayed affinities of 35.0 nM and 89.0 nM for CB1 and CB2 receptors, respectively, while the corresponding indazole 5F-AB-PINACA (16) was more potent displaying affinities in the low nanomolar range. We further investigated the 5Cl-pentyl derivative 5Cl-AB-PINACA (17), which showed comparable Ki values to 5F-AB-PINACA (16) at 4.06 nM for CB1 and 12.0 nM for CB2. The m-fluorobenzyl and the o-fluorobenzyl derivatives (18 and 19) showed similar affinities at the CB1 receptor, as also previously reported by Buchler et al. [37], with Ki values in the nanomolar range, and somewhat lower affinity for the CB2 receptor. AB-CHMINACA (20) displayed low nanomolar CB1 and CB2 affinity in agreement with previous results by Wiley et al. [41].
5F-ADB-PINACA isomer 2 (26) contains a structural isomer of isoleucinamide with a different side chain. This modification resulted in a slight decrease in affinities to CB1 and CB2 as compared to 5F-ADB-PINACA (23), the corresponding tert-leucinamide. Furthermore, tert-leucinamides, have been investigated which contain a tert-butyl group. The 5-fluoropentyl-substituted indole derivative 5F-ADBICA (21) showed nanomolar affinities with a Ki of 2.72 nM at CB1 and 1.83 nM at CB2 receptors. This was in agreement with data published by Banister et al. [42], who had reported similar EC50 values. We found the corresponding indazole derivative 23 to be slightly more potent with Ki values at 1.43 nM for CB1 and 0.694 nM for CB2. Banister et al. had determined a higher potency at CB1 with an EC50 value of 0.24 nM in their membrane potential assay, but a slightly higher EC50 value at CB2 (2.1 nM). The p-fluorobenzyl-substituted indazole ADB-FUBINACA (24) showed even lower Ki values of 0.360 nM for CB1 and 0.339 nM for CB2. The indole ADB-CHMICA (22) was substituted in the R2 position with a cyclohexylmethyl residue and showed a Ki value of 1.24 nM for the CB1 and 0.628 nM for the CB2 receptor. The corresponding indazole MAB-CHMINACA (25), which had been introduced by Buchler et al. [37], was even more potent with a Ki value of 0.333 nM for CB1 and 0.331 nM for CB2, which fits well with data reported by Buchler et al. for CB1 (no data for CB2 had been published by them).
PX-1 (27) and PX-2 (28) are phenylalaninamide derivatives, PX-1 (27) with an indole core and PX-2 (28) with an indazole core structure. PX-2 (28) showed a Ki value for the CB1 receptor of 127 nM and was thus significantly less potent than the corresponding tert-leucinamide derivative 5F-ADB-PINACA (23). The Ki value at CB2 (17.4 nM) was also higher than the Ki value of 0.694 nM determined for 5F-ADB-PINACA (23). Indole derivative PX-1 (27) displayed a Ki value of 485 nM for CB1, corresponding to a fourfold decrease in affinity as compared to the indazole PX-2 (28). The Ki value at CB2 (164 nM) was about tenfold higher. This confirms that the indazole ring system generally leads to a higher affinity as compared to the indole core structure.
APP-FUBINACA (29) and APP-CHMINACA (30) had been introduced by Buchler et al. [37]. Both are indazoles varying in position R2. The p-fluorobenzyl derivative APP-FUBINACA (29) showed potencies for both CB receptor subtypes of around 50 nM, while the corresponding cyclohexylmethyl derivative APP-CHMINACA (30) was more potent displaying Ki values of 9.81 nM for CB1 and 4.39 nM for CB2.
Instead of an amino acid residue, the R1 position has also been substituted with a cumyl moiety. These types of compounds were first described by Bowden and Williamson [43] and it has recently been found in illicit drug material. For all three investigated cumyl derivatives (31–33), we could demonstrate affinities in the low nanomolar range for the CB1 receptor. Bowden and Williamson had reported subnanomolar EC50 values in their functional assays using a homogeneous time-resolved fluorescence (HTRF)-based cAMP assay [43]. The indole derivatives Cumyl-PICA (31) and 5F-Cumyl-PICA (32) in our hands displayed potencies of around 25 nM for the CB2 receptor, while Cumyl-THPINACA (33) bearing a 4-tetrahydropyranylmethyl moiety (for R2) was more potent with a Ki value of 1.38 nM at the CB2 receptor, which was similar to its Ki value at the CB1 receptor.
The investigated series of compounds contained one member with a 3-oxycarbonyl linker: MO-CHMINACA (34), an indazole with a cyclohexylmethyl residue for R2 and a methoxycarbonyl-tert-leucine for R1. It displayed a Ki value of 10.4 nM at CB1 and 1.11 nM at CB2 receptors. The only other cyclohexylmethyl-substituted compound investigated by us was BB-22 (see our previous study [25]), which exhibited a Ki value of 0.217 nM for CB1 receptor; however it was substituted with a quinolone for R1 and contained an indole core.
Three 3-carbonylindoles (35–37) were studied. FUB-JWH-018 (35), substituted with a naphthyl residue for R1 and possessing a p-fluorobenzyl residue for R2 displayed similarly high nanomolar affinities like the previously studied naphthoyl indazoles THJ018 and THJ2201 [25]. MAM-2201 and EAM-2201, which were substituted with methyl or ethyl in the 4-position of the naphthoyl residue, had shown subnanomolar affinities [25]. Here we report F-2201 (36) and Cl-2201 (37), the respective 4-fluoro- and 4-chloro derivatives. Both displayed high affinities at 1–2 nM for both CB1 and CB2 receptors. The previously described alkyl-substituted naphthoyl derivatives had shown similar potencies (compare MAM-2201 and EAM-2201) [25]. The substitutions can be ranked in the following order of potency at CB1: ethyl > fluoro > chloro > methyl, while for CB2 it was: ethyl > methyl > fluoro ≈ chloro.
The indole derivative mepirapim (38) belongs to the 3-amido-substituted derivatives, featuring a 4-methylpiperazinyl residue for R1. Mepirapim (38) was originally identified by Uchiyama et al. [44] and has been found in "Spice" preparations. We determined an affinity of 2650 nM for the CB1 receptor and 1850 nM for the CB2 receptor. Therefore, it can be regarded as a rather weak CB receptor ligand.
We further investigated three structurally dissimilar compounds, 39–41, which contain a carbazole core substituted in position 3 with residues typically observed in position R1 of indazole- and indole-based compounds. EG-018 (39) and EG-2201 (40) feature a carbonyl linker connected to a naphthyl residue, whereas MDMB-CHMCZCA (41) is substituted with a methoxycarbonyl-tert-leucine residue through an amide linker. EG-018 (39) displayed low nanomolar affinities with Ki values of 7.17 nM for CB1 and of 2.27 nM for the CB2 receptor. EG-018 (39) can be compared to JWH-018 (3), which showed similar affinities. EG-2201 (40) was less potent at CB1 with a Ki value of 22.4 nM, but only slightly more potent at CB2 (Ki = 4.36 nM). MDMB-CHMCZCA (41) also displayed affinities in the low nanomolar range. The observed switch from indoles and indazoles to carbazoles can be interpreted as a reaction to the NpSG legislation and similar regulations in other countries that restricted the whole class of indoles and indazoles based on the known SARs. Recently, the synthetic cannabinoid Cumyl-PEGACLONE was identified as one of the first cannabimimetic compounds to circumvent these regulations; it consists of a γ-carboline, another new scaffold for cannabinoid receptor agonists [45]. Carbazoles (39–41) represent a further new scaffold which circumvents restrictions applied by many, especially European, countries by simply exchanging the well-established bicyclic core structures of indole or indazole for a tricyclic carbazole ring system.
We further investigated the benzimidazole derivative FUBIMINA (42), which had previously been described by Wiley et al. [41], and determined a Ki value of 502 nM at the CB1 receptor, which is in the same range as the reported Ki value of 296 nM, and a Ki value of 99.0 nM for the CB2 receptor, which is slightly higher than the reported value of 23.5 nM [41].
The presently investigated set of compounds complements our previous efforts to study the SARs of synthetic cannabinoids [25]. Of special interest is the observed scaffold hopping. Carbazole derivatives with a high affinity for CB receptors circumvent restriction by current law and display a new lead structure for CB receptor ligands. Further insight into the SARs is required to describe the potency profile of this compound class in more detail.
cAMP accumulation assays
As a next step, we investigated the compounds in cAMP accumulation assays, to obtain information on their functionality (Fig. 3). CB receptors are Gi protein-coupled and thus reduce the levels of cAMP in the cells upon activation. We applied the compounds at either 10 or 1 µM concentration depending on the Ki values measured in radioligand binding. If the Ki value was higher than 10 nM, we applied 10 µM of the compound in our assays; otherwise the lower concentration of 1 µM was assumed to be sufficient for maximal CB receptor activation. For comparison, we studied CP55,940 (1 µM), Δ9-THC (10 µM), and JWH-018 (1 µM) under the same conditions at concentrations at which they exert their maximal effects. The cAMP response of the full agonist CP55,940 (1 µM) was set at 100% receptor activation.

Receptor activation in cAMP accumulation assays. Receptor activation was normalized to the maximal effect observed with the full agonist CP55,940 (1 µM). Compounds were applied at 10 µM concentration in case their Ki value was ≥ 10 nM and at 1 µM concentration if their Ki value was < 10 nM. a Compounds 4–25; b compounds 26–33; c compounds 34–42
Moreover, we determined the EC50 values of MDMB-FUBINACA (12) by measuring full concentration inhibition curves. This compound had shown very low Ki values in radioligand binding assays indicating extremely high affinities, and in fact, the EC50 values determined in cAMP assays [EC50 values of 0.0641 nM (CB1) and 0.756 nM (CB2)] were in the same range as the Ki values measured in binding studies (see Fig. 4).

Pharmacological characterization of MDMB-FUBINACA (12). a Affinity of MDMB-FUBINACA (12) for the cannabinoid receptor CB1 determined in radioligand binding studies. b Receptor activation of the cannabinoid CB1 receptor by MDMB-FUBINACA (12) determined in cAMP accumulation assays. c Affinity of MDMB-FUBINACA (12) for the cannabinoid receptor CB2 determined in radioligand binding studies. d Receptor activation of the cannabinoid CB2 receptor by MDMB-FUBINACA (12) measured in cAMP accumulation assays
As can be seen in Fig. 3, almost all of the investigated compounds displayed agonistic behavior and showed high efficacy. Two compounds [5F-ADB (11) and APP-FUBINACA (29)] displayed a partial activation of the CB1 receptor at a concentration of 10 µM. At the CB2 receptor however, they were found to act as full agonists (Fig. 3). FUBIMINA (42) showed only partial activation of both CB1 and CB2 receptors at a concentration of 10 µM; the activation was similar to that of Δ9-THC, which is a partial agonist. Full receptor activation by FUBIMINA (42) might not have been observed, due to its low affinity for the receptors. In accordance with this, Wiley et al. [41] observed a micromolar EC50 of 2470 nM in [35S]GTPγS assays for FUBIMINA.
One compound completely lacked CB2 receptor activation: MDMB-CHMCZCA (41). However, MDMB-CHMCZCA (41) had shown high affinity for the CB2 receptor with a Ki value of 6.67 nM in radioligand binding studies. A higher concentration of MDMB-CHMCZCA (41) at 10 µM also failed to evoke an agonistic response (Fig. S3) on the CB2 receptor. Therefore, we investigated whether MDMB-CHMCZCA (41) might behave as a CB2 receptor antagonist. In Fig. 5d, the concentration-dependent response of MDMB-CHMCZCA (41) versus CP55,940 as an agonist (0.03 µM corresponding to its EC80 value) is shown. MDMB-CHMCZCA (41) displayed an IC50 value of 807 ± 137 nM under these conditions and clearly behaved as an antagonist at the CB2 receptor. The determined IC50 value in the cAMP assay was higher than the Ki value measured in radioligand binding studies. This might be due to the rather high concentration of CP55,940, that was applied, thus underestimating the inhibitory potency of 41. However, at the CB1 receptor MDMB-CHMCZCA (41) displayed agonistic behavior (Fig. 5b) with an EC50 value of 120 nM and showed full efficacy as compared to the full agonist CP55,940 (Fig. S4). Another carbazole derivative, EG-2201 (40) was investigated and found to induce agonistic behavior at both CB receptor subtypes. Its respective Ki and EC50 values were similar (CB1 Ki = 22.4 nM; EC50 = 15.6 nM; CB2 Ki = 4.36 nM and EC50 = 5.65 nM (see Fig. 6). It showed an efficacy of 94% at CB1 and 77% at the CB2 receptor as compared to the maximum response of the full agonist CP55,940 (see Fig. S4).

Pharmacological characterization of the carbazole derivative MDMB-CHMCZCA (41). a Affinity of MDMB-CHMCZCA (41) for the cannabinoid receptor CB1 determined in radioligand binding studies. b Receptor activation of the cannabinoid CB1 receptor by MDMB-CHMCZCA (41) determined in cAMP accumulation assays. c Affinity of MDMB-CHMCZCA (41) for the cannabinoid receptor CB2 determined in radioligand binding studies. d Inhibition of cannabinoid CB2 receptor activation induced by CP55,940 (0.03 µM) by MDMB-CHMCZCA (41) measured in cAMP accumulation assays

Pharmacological characterization of EG-2201 (40). a Affinity of EG-2201 (40) for the cannabinoid receptor CB1 in radioligand binding studies. b Receptor activation of the cannabinoid CB1 receptor by EG-2201 (40) determined in cAMP accumulation assays, as compared to the effect of forskolin (10 µM). c Affinity of EG-2201 (40) for the cannabinoid receptor CB2 determined in radioligand binding studies. d Receptor activation of the cannabinoid CB2 receptor by EG-2201 (40) measured in cAMP accumulation assays, as compared to the effect of forskolin (10 µM)
There are not many CB2 receptor antagonists known in the literature. As tool compounds, the inverse agonists AM-630 (43), an indole derivative, and SR-144,528 (44), a bornyl-substituted pyrazole, structurally related to the CB1 receptor inverse agonist rimonabant, are frequently employed. They are both selective for CB2 versus CB1 [46, 47]. This selectivity for the CB2 receptor might primarily be caused by the bulky lipophilic substituent attached to the heterocyclic core (R1 position). Compared to these structures (see Fig. 7), MDMB-CHMCZCA (41) represents a new class of CB2 receptor antagonists. It shares the bulky substitution of the known CB2 inverse agonists at R1, which covers a similar space as the bornyl substitutent of 44 (Fig. 7). Moreover, AM-630 (43) and SR-144,528 (44), share bulky lipophilic substituents at position 6 of the indole, or the methyl-chloro-phenyl moiety, respectively. MDMB-CHMCZCA (41) resembles these antagonists due to its voluminous tricyclic carbazole structure. While agonists induce a conformational change of the receptors leading to activation, competitive antagonists are often larger than agonists and just block the orthosteric binding site thereby preventing binding of the agonist.

Molecular docking studies
Recently, the crystal structure of the CB1 receptor was determined in both agonist- and antagonist-bound states with resolutions between 2.6 and 2.95 Å [33, 48, 49]. In a molecular docking study, we investigated possible binding poses and interactions of MDMB-FUBINACA (12), the most potent CB1 agonist of the present series (CB1 Ki = 0.0985 nM). For modeling of its interaction with MDMB-FUBINACA (12), we used the agonist-bound template structures. In both published templates, Δ9-THC-derived compounds were co-crystallized with the receptor. Here we compare these poses with the hypothetical poses obtained by the docking of MDMB-FUBINACA (12). The docking procedure was carried out using the Rosetta protein modeling suite of programs. The binding pose depicted by the largest cluster of low scoring models aligns the p-fluorobenzyl residue of MDMB-FUBINACA (12) with the alkyl side chain of the Δ9-THC derivative AM11542 (45) bound in the crystal structure (see Figs. 8, 9). This pose is regarded as plausible because the length of the p-fluorobenzyl residue of MDMB-FUBINACA (12) is of importance for CB1-selectivity versus CB2 and closely resembles the lipophilic side chain of 45. The co-crystallized agonist 45 showed a Ki value of 0.11 nM for the CB1 receptor [33], which is very similar to the affinity of MDMB-FUBINACA (12, K i 0.0985 nM). As shown in Figs. 8 and 9, the shape and size of both agonists as well as their lipophilicity and potential types of interaction aligned quite well. However, the template shows an interaction of serine-383 as a hydrogen bond donor to the phenolic group of the Δ9-THC-like compound. This was not observed in our model. Instead the oxygen atom of the ester function may participate in a hydrogen bond with histidine-178, an interaction that was not found for the co-crystallized compounds but could explain the equally high affinity of MDMB-FUBINACA (12) to the CB1 receptor observed in the present study. A plausible structural overlay of Δ9-THC derivative AM11542 (45) and MDMB-FUBINACA (12) is depicted in Fig. 9. Alternative binding poses were less often sampled and showed a superimposition of the tert-leucine methyl ester residue with the alkyl side chain (compare Fig. S1).

Docking of MDMB-FUBINACA (12) into the CB1 agonist state crystal structure reveals a plausible binding mode, in which the p-fluorobenzyl residue aligns with the alkyl side chain of the Δ9-THC-derived co-crystallized AM11542 (45, see Fig. 9)

Overlay of the CB1 receptor agonists MDMB-FUBINACA (12) and AM11542 (45)
Potency at the orphan cannabinoid-interacting GPCRs GPR18 and GPR55
The orphan receptors GPR18 and GPR55 can interact with certain natural and synthetic cannabinoids [26,27,28,29]. Recently, we found that some "Spice" constituents behaved as weak GPR55 antagonists [25]. Therefore, we investigated the new series of indole, indazole, benzimidazole and carbazole-derived structures in β-arrestin assays at GPR18 and GPR55 (Table 2). Most of the compounds were inactive. At GPR55, Cl-2201 (37) showed the highest antagonistic potency, tested versus the GPR55 agonist lysophosphatidylinositol (LPI, 1 µM), displaying an IC50 value of 7.12 µM. The fluorinated analogue F-2201 (36) was somewhat less potent with an IC50 value of 22.1 µM. Both of these compounds are derivatives of EAM-2201, which in our previous study had shown an IC50 value of 1.86 µM [25]. For the lipophilic substitutions, the following rank order of potency was observed: ethyl > methyl > chloro > fluoro. Therefore, it can be concluded that a lipophilic substitution in position 4 of the naphthyl residue was a requirement for GPR55 inhibition. The first amino acid-substituted derivatives to act as GPR55 antagonists are MO-CHMINACA (34) with an IC50 value of 9.29 µM and MDMB-CHMINACA (14) with an IC50 value of 10.3 µM. At GPR18 weak inhibitory potency was observed for MDMB-CHMICA (13), MO-CHMINACA (34) and MDMB-CHMCZCA (41).
These results indicate that the investigated series of CB receptor ligands is highly selective versus GPR18 and GPR55. None of the compounds was able to activate these orphan receptors. Some acted as antagonists at micromolar concentrations, but considerable efforts would be required to optimize these new lead structures to obtain potent GPR18- or GPR55-selective antagonists.
Conclusions
In this study, we continued to investigate the SARs of illicitly used constituents of "Spice" preparations. We investigated the affinities of a large series of compounds in radioligand binding assays and found MDMB-FUBINACA (12) belonging to the class of 3-amidoindazoles to be an extremely potent fully efficacious agonist showing picomolar affinities for CB1 (98.5 pM) and CB2 (130 pM) receptors. For this compound class severe side effects had been reported, as for example the “zombie outbreak” that was related to AMB-FUBINACA [50], a structurally related compound. The extremely high potency of these compounds might be one of the reasons for their severe side effects. The SARs, especially regarding the R2 residue, were consistent with the patterns observed in our previous study [25]. Lipophilic substituents had been introduced, e.g., a 5-fluoropentyl side chain, or a p-fluorobenzyl residue, which had similar properties as the pentyl side chain found in the JWH-compounds such as JWH-018. For MDMB-FUBINACA (12), we performed CB1 receptor docking studies and observed a pose comparable to Δ9-THC-derived compounds. In addition to the well-described group of alkylindoles and indazoles, we investigated a series of carbazoles, which showed single-digit nanomolar affinity at both CB receptor subtypes. One of these compounds, MDMB-CHMCZCA (41), unexpectedly turned out to be a full agonist at the CB1, but an antagonist at CB2 receptors, with K i values at CB1 of 5.75 nM and at CB2 of 6.67 nM, and EC50 values of 120 nM at CB1 and of 807 nM at CB2 receptors in cAMP accumulation assays. According to our knowledge, this combination of full CB1-agonistic and CB2-antagonistic activities is unique. Although CB2 receptor antagonists and inverse agonists have been studied for some time, their clinical utility is still under investigation. The expression of CB2 receptors in the immune system suggests immunomodulatory effects for CB2 receptor ligands. The group of carbazoles showed nanomolar affinities for the CB1 receptor and behaved as full agonists in cAMP accumulation assays. They circumvent the structural features described in the NpSG by scaffold hopping. This new class of synthetic cannabinoids needs to be further studied to fully investigate its SARs and potential for abuse. The present study may contribute to guiding future decisions on the restriction of carbazole-derived and related synthetic cannabinoids.
References
Lindigkeit R, Boehme A, Eiserloh I, Luebbecke M, Wiggermann M, Ernst L, Beuerle T (2009) Spice: a never ending story? Forensic Sci Int 191:58–63
European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) (2017) European drug report 2017: trends and developments. https://doi.org/10.2810/610791
European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) (2009) Thematic papers: understanding the ‘Spice’ phenomenon. https://doi.org/10.2810/27063
Davidson C, Opacka-Juffry J, Arevalo-Martin A, Garcia-Ovejero D, Molina-Holgado E, Molina-Holgado F (2017) Spicing up pharmacology. Adv Pharmacol 80:135–168
European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) (2015) European drug report 2015: trends and developments. https://doi.org/10.2810/314903
Fredriksson R, Lagerstrom MC, Lundin L-G, Schioth HB (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63:1256–1272
Ashton CH (2001) Pharmacology and effects of cannabis: a brief review. Br J Psychiatry 178:101–106
Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F (2008) CB2 receptors in the brain: role in central immune function. Br J Pharmacol 153:240–251
Pacher P, Mechoulam R (2011) Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50:193–211
Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11:563–583
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA (2010) International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62:588–631
Miller AM, Stella N (2008) CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol 153:299–308
Pertwee RG (2008) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215
Pertwee RG (2006) The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes 30 Suppl 1:13–18
Riederer AM, Campleman SL, Carlson RG, Boyer EW, Manini AF, Wax PM, Brent JA (2016) Acute poisonings from synthetic cannabinoids—50 U.S. Toxicology Investigators Consortium Registry Sites, 2010–2015. MMWR Morb Mortal Wkly Rep 65:692–695
U.S. Department of Justice, Drug Enforcement Administration (2018) Control division part 1308—schedules of controlled substances. 11 Schedule 1. https://www.deadversion.usdoj.gov/21cfr/cfr/1308/1308_11.htm. Accessed 6 March 2018
Bundesministerium der Justiz und für Verbraucherschutz (2016) Gesetz zur Bekämpfung der Verbreitung neuer psychoaktiver Stoffe. BGBl I Nr. 55
Bundesministerium für Verfassung, Reformen, Deregulierung und Justiz (2011) Bundesgesetz über den Schutz vor Gesundheitsgefahren im Zusammenhang mit Neuen Psychoaktiven Substanzen. BGBl. I Nr. 146/2011
Die Bundesversammlung der Schweizerischen Eidgenossenschaft (2011) Bundesgesetz über die Betäubungsmittel und die psychotropen Stoffe. SR 812.121
Huffman JW, Dai D, Martin BR, Compton DR (1994) Design, synthesis and pharmacology of cannabimimetic indoles. Bioorg Med Chem Lett 4:563–566
Huffman JW, Zengin G, Wu M-J, Lu J, Hynd G, Bushell K, Thompson ALS, Bushell S, Tartal C, Hurst DP, Reggio PH, Selley DE, Cassidy MP, Wiley JL, Martin BR (2005) Structure–activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid CB1 and CB2 receptors: steric and electronic effects of naphthoyl substituents. New highly selective CB2 receptor agonists. Bioorg Med Chem 13:89–112
Uchiyama N, Kikura-Hanajiri R, Kawahara N, Goda Y (2009) Identification of a cannabimimetic indole as a designer drug in a herbal product. Forensic Toxicol 27:61–66
Auwärter V, Dresen S, Weinmann W, Müller M, Pütz M, Ferreirós N (2009) ‘Spice’ and other herbal blends: harmless incense or cannabinoid designer drugs? J Mass Spectrom 44:832–837
Hess C, Schoeder CT, Pillaiyar T, Madea B, Müller CE (2016) Pharmacological evaluation of synthetic cannabinoids identified as constituents of spice. Forensic Toxicol 34:329–343
Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson N-O, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101
McHugh D, Page J, Dunn E, Bradshaw HB (2012) Δ9-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br J Pharmacol 165:2414–2424
Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, Abood ME (2009) Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem 284:29817–29827
Rempel V, Atzler K, Behrenswerth A, Karcz T, Schoeder C, Hinz S, Kaleta M, Thimm D, Kiec-Kononowicz K, Müller CE (2014) Bicyclic imidazole-4-one derivatives as: a new class of antagonists for the orphan G protein-coupled receptors GPR18 and GPR55. Med Chem Commun 5:632–649
Nordstedt C, Fredholm BB (1990) A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem 189:231–234
Bender BJ, Cisneros A 3rd, Duran AM, Finn JA, Fu D, Lokits AD, Mueller BK, Sangha AK, Sauer MF, Sevy AM, Sliwoski G, Sheehan JH, DiMaio F, Meiler J, Moretti R (2016) Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry 55:4748–4763
Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J (2010) Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry 49:2987–2998
Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho J-H, Han GW, Ding K, Li X, Liu H, Hanson MA, Zhao S, Bohn LM, Makriyannis A, Stevens RC, Liu Z-J (2017) Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547:468–471
Kothiwale S, Mendenhall JL, Meiler J (2015) BCL:conf: small molecule conformational sampling using a knowledge based rotamer library. J Chemin 7:47. https://doi.org/10.1186/A13321-015-0095-1
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Fujioka M, Omori N (2012) Subtleties in GPCR drug discovery: a medicinal chemistry perspective. Drug Discov Today 17:1133–1138
Buchler IP, Hayes MJ, Hedge SG, Hockerman SL, Jones DE, Kortum SW, Rico JG, Tenbrick RE, Wu KK (2009) Indazole derivatives. WO 2009/106982/A1
Banister SD, Longworth M, Kevin R, Sachdev S, Santiago M, Stuart J, Mack JBC, Glass M, McGregor IS, Connor M, Kassiou M (2016) Pharmacology of valinate and tert-leucinate synthetic cannabinoids 5F-AMBICA, 5F-AMB, 5F-ADB, AMB-FUBINACA, MDMB-FUBINACA, MDMB-CHMICA, and their analogues. ACS Chem Neurosci 7:1241–1254
Adamowicz P (2016) Fatal intoxication with synthetic cannabinoid MDMB-CHMICA. Forensic Sci Int 261:e5–e10
Bäckberg M, Tworek L, Beck O, Helander A (2017) Analytically confirmed intoxications involving MDMB-CHMICA from the STRIDA project. J Med Toxicol 13:52–60
Wiley JL, Marusich JA, Lefever TW, Antonazzo KR, Wallgren MT, Cortes RA, Patel PR, Grabenauer M, Moore KN, Thomas BF (2015) AB-CHMINACA, AB-PINACA, and FUBIMINA: affinity and potency of novel synthetic cannabinoids in producing Δ9-tetrahydrocannabinol-like effects in mice. J Pharmacol Exp Ther 354:328–339
Banister SD, Moir M, Stuart J, Kevin RC, Wood KE, Longworth M, Wilkinson SM, Beinat C, Buchanan AS, Glass M, Connor M, McGregor IS, Kassiou M (2015) Pharmacology of indole and indazole synthetic cannabinoid designer drugs AB-FUBINACA, ADB-FUBINACA, AB-PINACA, ADB-PINACA, 5F-AB-PINACA, 5F-ADB-PINACA, ADBICA, and 5F-ADBICA. ACS Chem Neurosci 6:1546–1559
Bowden MJ, Williamson JPB (2014) Cannabinoid compounds. WO2014/167530/A1
Uchiyama N, Shimokawa Y, Matsuda S, Kawamura M, Kikura-Hanajiri R, Goda Y (2014) Two new synthetic cannabinoids, AM-2201 benzimidazole analog (FUBIMINA) and (4-methylpiperazin-1-yl)(1-pentyl-1H-indol-3-yl)methanone (MEPIRAPIM), and three phenethylamine derivatives, 25H-NBOMe 3,4,5-trimethoxybenzyl analog, 25B-NBOMe, and 2C-N-NBOMe, identified in illegal products. Forensic Toxicol 32:105–115
Angerer V, Mogler L, Steitz J-P, Bisel P, Hess C, Schoeder CT, Müller CE, Huppertz LM, Westphal F, Schäper J, Auwärter V (2017) Structural characterization and pharmacological evaluation of the new synthetic cannabinoid CUMYL-PEGACLONE. Drug Test Anal. https://doi.org/10.1002/dta.2237
Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Brelière JC, Le Fur GL (1998) SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther 284:644–650
Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG (1999) Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol 126:665–672
Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, Rosenbaum DM (2016) High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540:602–606
Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, Laprairie RB, Stahl EL, Ho J-H, Zvonok N, Zhou H, Kufareva I, Wu B, Zhao Q, Hanson MA, Bohn LM, Makriyannis A, Stevens RC, Liu Z-J (2016) Crystal structure of the human cannabinoid receptor CB1. Cell 167:750–762
Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, Gerona R (2017) “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York. N Engl J Med 376:235–242
Longworth M, Banister SD, Boyd R, Kevin RC, Connor M, McGregor IS, Kassiou M (2017) Pharmacology of cumyl-carboxamide synthetic cannabinoid new psychoactive substances (NPS) CUMYL-BICA, CUMYL-PICA, CUMYL-5F-PICA, CUMYL-5F-PINACA, and their analogues. ACS Chem Neurosci 8:2159–2167
Rempel V, Volz N, Hinz S, Karcz T, Meliciani I, Nieger M, Wenzel W, Bräse S, Müller CE (2012) 7-Alkyl-3-benzylcoumarins: a versatile scaffold for the development of potent and selective cannabinoid receptor agonists and antagonists. J Med Chem 55:7967–7977
Rempel V, Fuchs A, Hinz S, Karcz T, Lehr M, Koetter U, Müller CE (2013) Magnolia extract, magnolol, and metabolites: activation of cannabinoid CB2 receptors and blockade of the related GPR55. ACS Med Chem Lett 4:41–45
Acknowledgments
We thank Marion Schneider for performing LC–MS analyses. CTS was supported by a BAYER Ph.D. fellowship; CEM and CTS are grateful for support by the Deutsche Forschungsgemeinschaft (Research Training Group GRK1873). We thank Prof. Volker Auwärter and Verena Angerer from the Institute of Forensic Medicine in Freiburg, Germany, for providing MDMB-CHMCZCA (41).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schoeder, C.T., Hess, C., Madea, B. et al. Pharmacological evaluation of new constituents of “Spice”: synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicol 36, 385–403 (2018). https://doi.org/10.1007/s11419-018-0415-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-018-0415-z