
Abstract
Differentiation among regioisomers of synthetic cannabinoids in forensic drug analysis is a crucial issue, since all isomers are not regulated by law. New equivalent analogs obtained via minor modification of their preexisting molecules keep on emerging. Isomers formed via substitutional exchange are also a cause for concern. This study is focused on the isomeric molecules that stem from minor modifications of 5F-PB-22. The analytical properties of these molecules and methods of differentiation are reported. Scan mode analysis using gas chromatography–electron ionization-mass spectrometry (GC–EI-MS) was performed using the authentic 5F-PB-22 standard, five regioisomeric quinolinyl ester indoles, and five regioisomeric isoquinolinyl ester indoles. Because it was not possible to separate 5F-PB-22 from the 5-hydroxyquinoline isomer using GC and all analytes showed similar EI mass spectra, liquid chromatography (LC)–tandem mass spectrometry analysis was performed. Using LC, a successful separation of 5F-PB-22 from all isomers could be achieved. Based on the electrospray ionization-mass spectra, the protonated molecular ion at m/z 377.2 was selected as the precursor ion for the regioisomeric and structural isomeric differentiation. Collision-induced dissociation provides relative intensity differences in the product ions among the isomers, enabling mass spectrometric differentiation of the isomers. To our knowledge, this is the first report on mass spectrometric differentiation of 5F-PB-22 and its ten isomers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An increasing number of new psychoactive designer drugs have emerged worldwide during the last decade [1]. Synthetic cannabinoids (SCs) constitute a major share of drug abuse in Japan as well as in European countries [2–5]. A survey of designer drugs available in the Japanese market was conducted under the leadership of the National Institute of Health Sciences (Tokyo). The identification of known SCs and the structural determination of newly emergent SCs were achieved using a combination of technologies such as hyphenated chromatographic procedures [gas chromatography–tandem mass spectrometry (GC–MS-MS) and liquid chromatography (LC)–MS-MS] and nuclear magnetic resonance spectroscopy [6–10].
Since new structurally modified analogs have been continuously emerging in the market, avoiding/reducing expansion of such illegal species is a never-ending challenge for governmental authorities, and the “cat-and-mouse game” between regulators and drug manufacturers is ongoing. For effective control of these new psychoactive substances, several legislative approaches have been implemented at the national level. The Ministry of Health, Labour and Welfare of Japan amended the Pharmaceutical Affairs Law in 2006 to implement a new regulatory category, called “Designated Substances (DSs)”. The most effective deterrent, a comprehensive regulatory system (generic scheduling) for naphthoylindole-type SCs and synthetic cathinones, was introduced to DSs in 2013. As of April 2016, 2343 compounds are controlled as DSs. The number of vendors on the street and the Internet has been reduced after the repeated expansion of DSs. On the contrary, such government control of designer SCs may ironically result in the continuing production of new designer drugs and an expansion of structurally related analogs in the clandestine market.
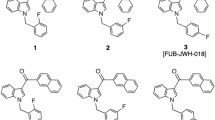
SCs bear the agonistic property for both cannabinoid receptors (CB1 and CB2), with various affinities and selectivities. The potential pharmacological activity on the CB1 receptors is responsible for the psychological effect and drug abuse [11, 12]. In the indole class SCs, indole-3-carboxylic acid ester derivatives with variation of the functional groups on the indole core constitute one of the representative SC groups in the DS list in Japan. The indole-3-carboxylates substituted by the 8-quinolinyl moiety (e.g., PB-22, 5F-PB-22, BB-22, FUB-PB-22) and the 1-naphthoyl moiety (e.g., FDU-PB-22) constitute the majority in this class (Fig. 1). 5F-PB-22 (also known as 5F-QUPIC) has been controlled as DS in 2013, and then has been re-categorized from DS to a narcotic in 2014 in Japan. Despite strict control under the Narcotics and Psychotropics Control Law, continuous survey of designer drugs in illegal markets by several prefectural governments, including Gifu, resulted in the ongoing detection of 5F-PB-22 in 2015 [13].

Chemical structures of synthetic cannabinoids having an indole core with 3-carboxylic acid ester
The next wave of SCs may include the structural variations resulting from positional isomerism. The fundamental studies on the positional isomer differentiation have only been performed using 1-alkyl-3-acylindoles, including JWH-081 and JWH-018, under the prospect of their future abuse [14–21]. However, there are no reports about ester- or amide-type SCs. We focused on 5F-PB-22, one of the ester-type SCs, and investigated the positional isomer differentiation of 5F-PB-22 involving five hydroxyquinoline isomers and five hydroxyisoquinoline isomers using GC–MS and LC–MS-MS.
Materials and methods
Reagents and chemicals
The structures of the 11 quinolinyl- and isoquinolinyl-substituted 1-(5-fluoropentyl)-1H-indole-3-carboxylates used in this study are shown in Figs. 1 and 2. Quinolin-8-yl 1-(5-fluoropentyl)-1H-indole-3-carboxylate (5F-PB-22: 1); its five hydroxyquinoline isomers [5F-PB-22 7-hydroxyquinoline isomer (7Q isomer, 2), 5F-PB-22 6-hydroxyquinoline isomer (6Q isomer, 3), 5F-PB-22 5-hydroxyquinoline isomer (5Q isomer, 4), 5F-PB-22 4-hydroxyquinoline isomer (4Q isomer, 5), 5F-PB-22 3-hydroxyquinoline isomer (3Q isomer, 6)]; and its five hydroxyisoquinoline isomers [5F-PB-22 8-hydroxyisoquinoline isomer (8IQ isomer, 7), 5F-PB-22 7-hydroxyisoquinoline isomer (7IQ isomer, 8), 5F-PB-22 6-hydroxyisoquinoline isomer (6IQ isomer, 9), 5F-PB-22 5-hydroxyisoquinoline isomer (5IQ isomer, 10), and 5F-PB-22 4-hydroxyisoquinoline isomer (4IQ isomer, 11)] were purchased from Cayman Chemical (Ann Arbor, MI USA). The stock standard solutions of these compounds were prepared at a concentration of 1 mg/mL (in acetonitrile) and were stored at −20 °C until analysis. These solutions were diluted in acetonitrile to appropriate concentrations prior to analysis. All other chemicals and solvents were of analytical reagent grade or LC–MS grade. Twelve herbal products (GP-2015-01 to GP-2015-12) were purchased via the Internet in March 2015 in Japan. All the products contained approximately 3–4 g of mixed dried plants.

Chemical structures of ten isomers of 5F-PB-22 (1). 2, 5F-PB-22 7-hydroxyquinoline isomer (7Q isomer); 3, 5F-PB-22 6-hydroxyquinoline isomer (6Q isomer); 4, 5F-PB-22 5-hydroxyquinoline isomer (5Q isomer); 5, 5F-PB-22 4-hydroxyquinoline isomer (4Q isomer); 6, 5F-PB-22 3-hydroxyquinoline isomer (3Q isomer); 7, 5F-PB-22 8-hydroxyisoquinoline isomer (8IQ isomer); 8, 5F-PB-22 7-hydroxyisoquinoline isomer (7IQ isomer); 9, 5F-PB-22 6-hydroxyisoquinoline isomer (6IQ isomer); 10, 5F-PB-22 5-hydroxyisoquinoline isomer (5IQ isomer); and 11, 5F-PB-22 4-hydroxyisoquinoline isomer (4IQ isomer)
Preparation of sample solution
The herbal-type product (30 mg) was crushed to a powder and extracted with 3 mL of methanol or acetonitrile under ultrasonication for 10 min. After centrifugation (5 min, 3000 rpm), the supernatant solution was passed through a membrane filter (Millex-LH 0.45 μm filter unit; Merck Millipore, Darmstadt, Germany). Moreover, if necessary, the solution was diluted with methanol or acetonitrile to a suitable concentration before instrumental analysis.
Analytical methods
GC–MS analysis was performed using a TRACE 1310 GC and ISQ LT (Thermo Fisher Scientific, Waltham, MA, USA). Samples were separated using a DB-5MS capillary column (30 m × 0.25 mm i.d.; 0.25 μm film thickness; Agilent Technologies, Santa Clara, CA, USA) with helium as the carrier gas, at a constant flow rate of 1.0 mL/min. The oven temperature was programmed as follows: held at 50 °C for 1 min, linearly ramped at 10 °C/min to 310 °C, and held at 310 °C for 12 min. The injector temperature was set to 250 °C, and the injection volume was 1 μL (splitless mode). The GC interface and ion-source temperature were maintained at 280 and 250 °C, respectively. Ionization was performed in the electron ionization (EI) mode at 70 eV. A mass spectral library search was performed using the Searchable Mass Spectral Library Version 2.2 downloaded from the provider’s website [22].
GC–MS-MS was performed using an Agilent 7890A/7000 GC/MS Triple Quad (Agilent Technologies). The positive EI mode was employed. Samples were introduced via a DB-5MS + DG capillary column (30 m × 0.25 mm i.d.; 0.25 μm film thickness with 10 m DuraGuard; Agilent Technologies). The analytical conditions were similar to those in the GC–MS analysis. Product ion spectra were obtained using N2 as the collision gas (1.5 mL/min) by varying the collision energy (CE) from 5 to 35 V, in units of 10 V.
LC equipped with a photodiode array (PDA) detector analysis was performed on an Agilent Technologies liquid chromatograph (1100 series). Chromatographic separation was carried out on an Atlantis T3 column (150 × 4.6 mm i.d., particle size 5 μm; Waters, Milford, MA, USA). The mobile phase comprised (A) 0.1 % formic acid in water and (B) 0.1 % formic acid in acetonitrile. The column was maintained at 40 °C, and the gradient program was as follows: linear gradient of B from 5 to 90 % from 0 to 45 min and isocratic elution of 90 % B from 45 to 60 min. The flow rate and the injection volume were 1.0 mL/min and 10 μL, respectively.
LC–MS analysis was performed on an Agilent 1100 Series LC/MSD-equipped with electrospray ionization (ESI) and an Atlantis T3 column (150 × 2.1 mm i.d., particle size 5 μm; Waters). The mobile phase and column temperature were similar to those in LC–PDA. The flow rate and injection volume were 0.2 mL/min and 2 μL, respectively.
LC–MS-MS analysis was performed on a 1200 Series LC (Agilent Technologies) and a 4000 QTRAP (AB Sciex, Framingham, MA, USA) equipped with ESI setup and an Atlantis T3 column (150 × 2.1 mm i.d., particle size 5 μm; Waters). The mobile phase and column temperature were the same as those in LC–PDA. The flow rate was 0.2 mL/min, and the injection volume was 1 μL. The MS was operated in the positive mode. The ion spray voltage was 5500 V, and the desolvation temperature was 600 °C. Product ion spectra were obtained at CEs of 10, 20, 30, and 35 V.
Results and discussion
Identification of 5F-PB-22 (1) from herbal products
During the course of the Gifu prefectural governmental program to suppress drug abuse in 2015, 12 unknown herbal-type products (GP-2015-01 to GP-2015-12) were purchased on the Internet, and comprehensive analyses were performed using GC–MS, LC–PDA, and LC–MS. Identification of the detected peaks from each analytical technique was first achieved using the available database and finally by comparison of the data obtained from an authentic standard. This resulted in the identification and quantification of a narcotic 5F-PB-22 from two herbal products, GP-2015-04 (65 mg/g) and GP-2015-05 (82 mg/g), respectively named LAST Kou No. 1 and No. 2 [13]. LC–PDA and LC–MS analyses of the two herbal products showed a peak that had retention time (Rt), UV spectrum, and mass spectrum coincident with those of the reference standard of 5F-PB-22. The results of GC–MS analysis obtained from GP-2015-05 are shown in Fig. S1. The examination directed toward detections of the quinolinyl- and isoquinolinyl-substituted regioisomers of 5F-PB-22 were also conducted thoroughly by all the procedures described in the following sections. However, the authors failed to detect the isomer(s) from the authentic herbal products.
It is well known that SCs having a heat-unstable core skeleton of (1H-indole)-3-carboxylate tend to give analytical artifacts, resulting from the transesterification reaction, when they are analyzed by GC–MS in the presence of methanol or ethanol [23, 24]. The degradation and esterification occur in the injection port of the GC instrument. We also demonstrated the detection of decomposed 5F-PB-22 during the analysis of GP-2015-04 and GP-2015-05. Intense peaks corresponding to the methyl ester (methyl 1-(5-fluoropentyl)-(1H-indole)-3-carboxylate) (Rt = 20.91 min), 8-hydroxyquinoline (Rt = 11.51 min) and a small peak corresponding to the intact 5F-PB-22 (Rt = 31.58 min) were observed in the total ion chromatogram in the presence of methanol (Fig. S1a). To avoid a low sensitivity for the targeted analysis of SCs with (1H-indole)-3-carboxylate, acetonitrile was adopted as the solvent for extraction of all herbal products and in the preparation of standard solutions of authentic samples. This resulted in a higher sensitivity for the target molecule, 5F-PB-22 (Fig. S1b).
GC–MS and GC–MS-MS analyses
EI mass spectra of the reference standards of 5F-PB-22 (1) and its isomers (2–11) are shown in Fig. S2. These EI mass spectra were acquired individually following sample injection using GC–MS. 5F-PB-22 (1) and all ten isomers (2–11) equally showed a small molecular ion peaks at m/z 376. The ion at m/z 232 ([M–144]+: C14H15FNO+) occurred as the base peak for each of the 11 compounds and represents the α-cleavage of a carbonyl group, i.e., the loss of a hydroxyquinoline radical from the molecular ion to afford an N-1-(5-fluoropentyl)-indolylacylium ion (Fig. 3). Additionally, all the compounds showed characteristic fragmentation patterns for 3-acylindoles, resulting in fragments at m/z 144 and 116 of similar relative intensities. 5F-PB-22 (1) and its ten isomers (2–11) were not distinguishable based on their EI mass spectra. Structural isomeric differentiation was also investigated using the collision-induced dissociation (CID) of ions for the molecular ion peak (m/z 376) and the base peak (m/z 144). The product ion spectra of m/z 376 for 5F-PB-22 (1) and its ten isomers (2–11) had a very low sensitivity due to an insufficient precursor ion; most molecular ions were disrupted by α-cleavage with EI. It was estimated that the precursor ion at m/z 144 was attributed to C9H6NO+, not only due to the indolylacylium ion [18–21, 25, 26], but also due to the positively charged quinolinol moiety present in 2–6 [10, 24] or the isoquinolinol moiety in 7–11. However, all the compounds (1–11) yielded identical product ion spectra of m/z 144 (Fig. S3), which suggested that the abundance of the m/z 144 ion was due to the 3-acylindole ring rather than the hydroxy(iso)quinoline ring. The differentiation of 5F-PB-22 (1) and its ten isomers (2–11), based on the product ion spectra acquired from GC–MS–MS, was unfortunately unsuccessful.

Putative fragmentation scheme for 5F-PB-22 and its isomers following electron ionization
The two characteristic fragment ions at m/z 144 (C9H6NO+) and 116 (C8H6N+) arose from the elimination of olefin (5-fluoro-1-pentene) from the N-1-(5-fluoropentyl)-indolylacylium ion and the subsequent loss of CO [25]. The olefin elimination occurred with rearrangement of an H-atom from the m/z 232 ion bearing a positively charged N-atom resonance, as illustrated in Fig. 3 [20]. The reaction requires the existence of an N-substituent with an H-atom at a β position relative to the indole core. Previous reports showed supporting evidence for the elimination mechanism in the EI mass spectra from two quinolin-8-yl 1H-indole-3-carboxylates, BB-22 [10] and FUB-PB-22 [9] (Fig. 1). The former has a cyclohexylmethyl group with a β H-atom and yields the m/z 144 ion due to elimination of the substituent. The latter, substituted with a fluorobenzyl group without such an H-atom, did not show any m/z 144 ion. This evidence further reinforces the inference that the m/z 144 ion is formed due to the indolylacylium ion alone and stems from the N-1-(5-fluoropentyl)-indolylacylium ion at m/z 232. The EI mass spectra of some synthetically prepared degradation products of PB-22, without a quinolinyl group, have been reported to show the intense product ion at m/z 144 [24], which further supports the conclusion that the m/z 144 ion is formed from the indolylacylium ions.
GC studies
The GC Rts of 5F-PB-22 (1) and its ten isomers (2–11) are shown in Fig. 4. Under the present experimental conditions, the chromatography yielded excellent resolution between 1 and each regioisomer (2–11), except for 5Q (4) and 4Q (5) isomers. The first compound that eluted from the column was 5Q isomer (4) (Rt = 31.60 min), followed in order by 1 (Rt = 31.63 min), 5 (Rt = 31.69 min), and 5IQ isomer (10) (Rt = 31.91 min). The other compounds (2, 3, 6–9, and 11) were detected in the region Rt = 32.30–33.45 min. 5Q isomer (4) and 4Q isomer (5) were very close to 1 in retention time. Analysis of a mixture of 1 and 4 at the same concentration resulted in a single and symmetric peak (Fig. S4a). A mixture of 1 and 5 showed a split in the peak (Fig. S4b). These results show that the retention properties of 4 and 5 are extremely similar to 1 and suggest that further combined analysis using hyphenated methods must be performed for these isomers.

Gas chromatographic retention times for 5F-PB-22 and its isomers
GC shows that six quinolinyl 1-(5-fluoropentyl)-1H-indole-3-carboxylates (1–6) can be grouped into two (Group A: 4, 1, and 5; Group B: 6, 3, and 2) categories according to their retention times and structural features (Fig. 4). It is interesting that compounds from group A bear a common structural feature in the quinolinyl moiety, where the O-atom is substituted closest to the C-atoms, C4a or C8a, of the quinoline ring. These three compounds have in common the close intramolecular relationship between the two indole substituents, where a quinoline ring and indole ring situated angularly form more compact structures compared to the compounds from group B. The 5-fluoropentyl and quinolinyl groups are more crowded in these three compounds, suggesting that interactions between the two groups minimize the retention time relative to the other isomers in group B. Compounds of group B, on the other hand, also bear a common structural feature in the quinolinyl moiety, where the O-atom is substituted farthest from C4a or C8a of the quinoline ring. The quinoline ring and indole ring are positioned linearly to form an extended structure that would contribute to the similar physical properties observed in compounds from Group B. From the conformational search, performed using Pcmodel with the MMFF94 force field (Serena Software, Bloomington, IN, USA), energy-minimized structures were obtained for compounds 1–6. The results for 5F-PB-22 (1) and 7Q isomer (2) are shown in Fig. 5, and indicate a closer alignment of the quinoline ring and 5-fluoropentyl chain on the indole ring for 1 and a more extended geometry for 2. These observations support the above-mentioned arguments on the structural features of the molecules. The same arguments apply for retention properties and the structural features of five isoquinolinyl 1-(5-fluoropentyl)-1H-indole-3-carboxylates (7–11), which show a relatively shorter retention time in the case of group A (10, 11, and 7) with compact structural features, and an increased retention time for group B (8 and 9) with extended structural features (Fig. 4).

Energy-minimized structures for 1 5F-PB-22 and 2 7Q isomer based on conformational search using Pcmodel with the MMFF94 force field
LC–PDA and LC–MS analyses
LC analyses, using LC–MS and LC–PDA, were employed to develop suitable procedures for the separation of 5F-PB-22 (1) and each of the ten isomers (2–11). The chromatogram from LC–MS showed that the peaks corresponding to 5F-PB-22 (1) and ten isomers (2–11) were in the region of Rt = 29.11–40.49 min (Fig. 6). LC analysis facilitated the complete separation of 1 (Rt = 39.96 min), 4 (Rt = 36.36 min), and 5 (Rt = 34.43 min), although they overlapped during GC analysis. The compounds studied exhibited incomparable UV absorption spectra with similarity over the range of 200–350 nm (Fig. S5). All the isomeric compounds (2–11) exhibited mass spectra similar to 5F-PB-22 (1), which showed a quasi-molecular ion at m/z 377 and a fragment ion at m/z 232, on LC–MS analysis (data not shown).

Liquid chromatographic retention times for 5F-PB-22 and its isomers
LC–MS–MS analysis
Mass spectrometric differentiation among 5F-PB-22 (1) and its ten isomers (2–11) was attempted using CID by targeting the protonated molecular ion at m/z 377.2 as the precursor ion. The analyses were performed at CEs of 10, 20, 30, and 35 V using an LC–MS-MS in the ESI mode. The product ion spectra for 5F-PB-22 (1) and five Q isomers (2–7) at CEs of 10 and 20 V are shown in Fig. 7. The major product ion was only at m/z 232, and the other fragment ion was hardly observed. The only difference among the product ion spectra of the ten isomers was their relative intensities of the product ion at m/z 232, which was used for isomer differentiation. The relative intensities are summarized in Table 1, based on the precursor ion at m/z 377 and the product ion at m/z 232. At a CE of 10 V, 5F-PB-22 (1) and 4Q isomer (5) produced the product ion at m/z 232 with relative intensities of 57 and 100 %, respectively. For nine isomers (2–4 and 6–11), other than 4Q isomer (5), the product ions at m/z 232 were not detected or were detected at very low relative intensities (<10 %). At a CE of 20 V, 5F-PB-22 (1) showed ions at m/z 232 and 377 with relative intensities of 100 and 11 %, respectively. The precursor ion at m/z 377 was not detected at CEs of 30 and 35 V. The relative intensities differed from isomer to isomer depending on CE. This reflects the difference in energy required for α-cleavage that depends on the molecular structure of each isomer. Based on a relative-intensity comparison among 5F-PB-22 (1) and its five Q isomers (2–6), the energy required for α-cleavage is estimated as follows: 6Q isomer > 3Q isomer > 5Q isomer > 7Q isomer > 5F-PB-22 > 4Q isomer. As a result, isomer differentiation was possible based on the product ion spectra at several steps of CE using LC–MS-MS. It would be effective to confirm the conformity of the relative intensity with a reference standard using LC–MS-MS for preventing the misidentification of these isomers.

Product ion spectra of precursor ion at m/z 377 for 5F-PB-22 and five Q isomers at CEs of 10 V (left) and 20 V (right) on LC–MS-MS. a 5F-PB-22, b 7Q isomer, c 6Q isomer, d 5Q isomer, e 4Q isomer, and f 3Q isomer
Conclusions
We conducted hyphenated chromatographic analysis for regioisomers of hydroxyquinolinyl ester indoles and hydroxyisoquinolinyl ester indoles of 5F-PB-22. All isomers were indistinguishable from 5F-PB-22 by EI–MS and EI–MS–MS analyses coupled to a GC system, due to their superimposable spectra. SCs having a carboxy ester group at the C-3 position of indole hardly generate a detectable ion stemming from quinolinyl- or isoquinolinyl-moiety, which renders structural differentiation difficult. However, all isomers were distinguishable from 5F-PB-22 by combining results from GC and LC separations. Although the GC system showed close retention among 5F-PB-22 (1), 5Q isomer (4), and 4Q isomer (5), they were completely resolved in the LC system using ODS adsorbent. The elution order in the GC system showed that the stereochemically extended isomers have a higher affinity for the capillary column. While the UV and ESI mass spectra obtained from the LC system were also not suitable for structural differentiation due to their close similarity, ESI-MS-MS analysis enabled the differentiation of the isomers by means of differences in relative ion intensity for the product ion at m/z 232, derived from the protonated quasi-molecular ion at m/z 377. The recent research trend regarding the structural differentiation of SCs, supervised by Zaitsu [14, 16] and Clark [15, 17–20], is focused on 1-alkyl-3-acylindoles. The series of isomers in the GC system display significant fragment ions stemming from the isomeric acyl cation at equivalent masses, with some differences in relative abundance of these ions, which is the biggest advantage of using EI-MS for this purpose. On the other hand, 1-alkyl-(1H-indole)-3-carboxylates can hardly generate a detectable ion from isomeric moieties, e.g., quinolinyl ester and isoquinolinyl ester conducted in the current study, hindering the development of an analytical procedure based on EI-MS for such compounds. This problem is encountered not only in case of EI-MS-aided structural differentiation of other regioisomeric quinolinyl or isoquinolinyl ester indoles of PB-22, BB-22, and FUB-PB-22, but also for indazole derivatives, e.g., 5F-NPB-22, NPB-22, and FUB-NPB-22. In addition to the differentiation procedure by resolution in GC and LC retention, an alternate procedure is required for future forensic examinations. The potential procedure aimed toward tackling this challenging subject involves the utilization of product ion spectra in the ESI mode, which would contribute to the analysis of forthcoming designated substances.
References
Underwood E (2015) A new drug war. Science 347:469–473
Kikura-Hanajiri R (2016) New designer drugs in Japan. In: Victor RP (ed) Neuropathology of drug addictions and substance misuse, vol 2. Academic Press, Cambridge, pp 1055–1065
United Nations Office on Drugs and Crime (2015) Fifteen novel synthetic cannabinoids reported to UNODC EWA since the beginning of the year. http://www.unodc.org/LSS/Announcement/Details/d05ee4c0-c835-4055-b042-df0a15f16f49. Accessed June 2016
European Monitoring Centre for Drugs and Drug Addiction (2015) EMCDDA–Europol 2014 annual report on the implementation of Council Decision 2005/387/JHA, Implementation reports, Publications Office of the European Union, Luxembourg. http://www.emcdda.europa.eu/system/files/publications/1018/TDAN15001ENN.pdf. Accessed June 2016
European Monitoring Centre for Drugs and Drug Addiction (2015) European drug report 2015: trends and developments. Publications Office of the European Union, Luxembourg. http://www.emcdda.europa.eu/publications/edr/trends-developments/2015. Accessed June 2016
Uchiyama N, Kikura-Hanajiri R, Hakamatsuka T (2016) A phenethylamine derivative 2-(4-iodo-2,5-dimethoxyphenyl)-N-[(3,4-methylenedioxyphenyl)methyl]ethanamine (25I-NB34MD) and a piperazine derivative 1-(3,4-difluoromethylenedioxybenzyl)piperazine (DF-MDBP), newly detected in illicit products. Forensic Toxicol 34:166–173
Uchiyama N, Asakawa K, Kikura-Hanajiri R, Tsutsumi T, Hakamatsuka T (2015) A new pyrazole-carboxamide type synthetic cannabinoid AB-CHFUPYCA [N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-3-(4-fluorophenyl)-1H-pyrazole-5-carboxamide] identified in illegal products. Forensic Toxicol 33:367–373
Uchiyama N, Shimokawa Y, Kikura-Hanajiri R, Demizu Y, Goda Y, Hakamatsuka T (2015) A synthetic cannabinoid FDU-NNEI, two 2H-indazole isomers of synthetic cannabinoids AB-CHMINACA and NNEI indazole analog (MN-18), a phenethylamine derivative N-OH-EDMA, and a cathinone derivative dimethoxy-α-PHP, newly identified in illegal products. Forensic Toxicol 33:244–259
Uchiyama N, Shimokawa Y, Kawamura M, Kikura-Hanajiri R, Hakamatsuka T (2014) Chemical analysis of a benzofuran derivative, 2-(2-ethylaminopropyl)benzofuran (2-EAPB), eight synthetic cannabinoids, five cathinone derivatives, and five other designer drugs newly detected in illegal products. Forensic Toxicol 32:266–281
Uchiyama N, Matsuda S, Kawamura M, Kikura-Hanajiri R, Goda Y (2013) Two new-type cannabimimetic quinolinyl carboxylates, QUPIC and QUCHIC, two new cannabimimetic carboxamide derivatives, ADB-FUBINACA and ADBICA, and five synthetic cannabinoids detected with a thiophene derivative α-PVT and an opioid receptor agonist AH-7921 identified in illegal products. Forensic Toxicol 31:223–240
Aung MM, Griffin G, Huffman JW, Wu M, Keel C, Yang B, Showalter VM, Abood ME, Martin BR (2000) Influence of the N-1 alkyl chain length of cannabimimetic indoles upon CB(1) and CB(2) receptor binding. Drug Alcohol Depend 60:133–140
Wiley JL, Compton DR, Dai D, Lainton JA, Phillips M, Huffman JW, Martin BR (1998) Structure-activity relationships of indole- and pyrrole-derived cannabinoids. J Pharmacol Exp Ther 285:995–1004
Gifu Prefectural Government (2015) Media release, 11th June. http://www.pref.gifu.lg.jp/kodomo/iryo/yakuji/11224/dappouhahbu.data/kisyahappyou1506101.pdf. Accessed May 2016
Kusano M, Yamanaka M, Zaitsu K, Nakayama H, Nakajima J, Moriyasu T, Tsuchihashi H, Ishii A (2016) Regioisomeric differentiation of the alkyl-substituted synthetic cannabinoids JWH-122 and JWH-210 by GC-EI-MS/MS. Forensic Toxicol 34:304–315
Abdel-Hay KM, De Ruiter J, Smith F, Alsegiani AS, Thaxton-Weissenfluh A, Clark CR (2016) GC–MS differentiation of the six regioisomeric dimethoxybenzoyl-1-pentylindoles: isomeric cannabinoid substances. J Pharm Biomed Anal 125:360–368
Kusano M, Zaitsu K, Nakayama H, Nakajima J, Hisatsune K, Moriyasu T, Matsuta S, Katagi M, Tsuchihashi H, Ishii A (2015) Positional isomer differentiation of synthetic cannabinoid JWH-081 by GC–MS/MS. J Mass Spectrom 50:586–591
Thaxton A, Belal TS, Smith F, DeRuiter J, Abdel-Hay KM, Clark CR (2015) GC–MS studies on the six naphthoyl-substituted 1-N-pentyl-indoles: JWH-018 and five regioisomeric equivalents. Forensic Sci Int 252:107–113
Abdel-Hay KM, DeRuiter J, Smith F, Belal TS, Clark CR (2015) GC–MS analysis of the regioisomeric methoxy- and methyl-benzoyl-1-pentylindoles: isomeric synthetic cannabinoids. Sci Justice 55:291–298
DeRuiter J, Smith FT, Abdel-Hay K, Clark CR (2014) Analytical differentiation of 1-alkyl-3-acylindoles and 1-acyl-3-alkylindoles: isomeric synthetic cannabinoids. Anal Chem 86:3801–3808
Smith FT, DeRuiter J, Abdel-Hay K, Clark CR (2014) GC–MS and FTIR evaluation of the six benzoyl-substituted-1-pentylindoles: isomeric synthetic cannabinoids. Talanta 129:171–182
Shevyrin V, Melkozerov V, Nevero A, Eltsov O, Shafran Y (2013) Analytical characterization of some synthetic cannabinoids, derivatives of indole-3-carboxylic acid. Forensic Sci Int 232:1–10
Scientific Working Group for the Analysis of Seized Drugs (2015) http://www.swgdrug.org/. Accessed April 2015
Blakey K, Boyd S, Atkinson S, Wolf J, Slottje PM, Goodchild K, McGowan J (2016) Identification of the novel synthetic cannabimimetic 8-quinolinyl 4-methyl-3-(1-piperidinylsulfonyl)benzoate (QMPSB) and other designer drugs in herbal incense. Forensic Sci Int 260:40–53
Tsujikawa K, Yamamuro T, Kuwayama K, Kanamori T, Iwata YT, Inoue H (2014) Thermal degradation of a new synthetic cannabinoid QUPIC during analysis by gas chromatography–mass spectrometry. Forensic Toxicol 32:201–207
Wohlfarth A, Gandhi AS, Pang S, Zhu M, Scheidweiler KB, Huestis MA (2014) Metabolism of synthetic cannabinoids PB-22 and its 5-fluoro analog, 5F-PB-22, by human hepatocyte incubation and high-resolution mass spectrometry. Anal Bioanal Chem 406:1763–1780
Takayama T, Suzuki M, Todoroki K, Inoue K, Min JZ, Kikura-Hanajiri R, Goda Y, Toyo’oka T (2014) UPLC/ESI-MS/MS-based determination of metabolism of several new illicit drugs, ADB-FUBINACA, AB-FUBINACA, AB-PINACA, QUPIC, 5F-QUPIC and α-PVT, by human liver microsome. Biomed Chromatogr 28:831–838
Acknowledgments
This work was funded by the domestically programmed grant for the regional society from the Gifu Prefectural Research Institute for Health and Environmental Sciences. The study was supported by the Health and Labour Sciences Research Grants 2015 and 2016 to K. Kitaichi (Research on Regulatory Science of Pharmaceuticals and Medical Devices, No. 27170401). A portion of this work was supported by the governmental program on a survey of designer drugs in the illegal drug market, supervised by Gifu Prefectural Government, Japan. We acknowledge the Gifu Regional Consortium on the Development of Analytical Procedures for Legal Highs operated by Gifu Pharmaceutical University and the Gifu Prefectural Research Institute for Health and Environmental Sciences for overseeing the experimental protocols.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no financial or other relations that could lead to a conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kohyama, E., Chikumoto, T., Tada, H. et al. Analytical differentiation of quinolinyl- and isoquinolinyl-substituted 1-(5-fluoropentyl)-1H-indole-3-carboxylates: 5F-PB-22 and its ten isomers. Forensic Toxicol 35, 56–65 (2017). https://doi.org/10.1007/s11419-016-0334-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-016-0334-9