
Abstract
Purpose
To explore the mechanisms in the deposition and release of phosphorus (P) in the sediment of a shallow eutrophic lake using preserved samples, we investigated the vertical and temporal changes in P, manganese (Mn), sulfur (S), iron (Fe), aluminum (Al), calcium (Ca), and magnesium (Mg) in the sediment samples and the phosphate in the sediment pore water samples over a period of 6 years.
Materials and methods
The upper 15 cm of sediment from Lake Kasumigaura in Japan was collected monthly from 2003 to 2008 from the center of the lake. Sediment cores were divided into seven depth segments and were acid-digested for an elemental analysis via inductively coupled plasma atomic emission spectroscopy. Phosphate concentrations of the sediment pore water were determined using the molybdenum blue method. A multiple regression analysis was conducted by setting the P content as the response variable and Mn, S, Fe, Al, Ca, and Mg as explanatory variables.
Results and discussion
The results of the multiple regression analysis demonstrated that P co-precipitates with Fe and Al oxides and accumulates on the sediment surface. The vertical distributions of Mn and S suggest that Mn reduction occurs within the 0–1-cm-depth layer of the sediment and that iron sulfide is actively formed in the 6–10-cm-depth layer of the sediment. These findings imply that the layer in which ferric oxides are reduced to ferrous ions is present near the 1–6-cm-depth layer of the sediment. This layer corresponds to the layer in which the maximum phosphate concentration of the sediment pore water often occurred (the 2–6-cm-depth layer). These results indicate that vertical distributions of mineral elements are useful for assessing P dynamics in sediments.
Conclusions
The lake sediments record the dynamics of P in the sediment. Our analytical approach using long-term observation data demonstrated that the accumulation and release of P associated with a change in the redox state can be assessed based on the vertical distributions of mineral elements in the lake sediments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Sediments generally act as a sink for phosphorus (P) in lakes (Golterman 1995). Phosphate in the water column co-precipitates with aluminum (Al), iron (Fe), and manganese (Mn) oxides; these co-precipitated phosphates accumulate on sediment surfaces as Al-bound P, Fe-bound P, and Mn-bound P, respectively (Ruttenberg 1992; Olila et al. 1995; Welch and Cooke 1995; Søndergaard et al. 2003; Wang et al. 2005; Hupfer et al. 2007). Authigenic carbonate fluorapatite (calcium (Ca)-bound P) also accumulates and acts as a sink for P in sediments (Ruttenberg 2003). In addition to these inorganic P sinks, suspended particles such as bacteria, detritus, and phytoplankton are also deposited and accumulated on surface sediments as biogenic P in lakes (Goedkoop and Pettersson 2000). To trace P originating from phytoplankton cells in sediments, the magnesium (Mg) content in sediments can be used as an indicator. This is because all photosynthetic primary producers include chlorophyll a, which consists of a group of Mg coordination complexes of cyclic tetrapyrroles. These findings indicate that the deposition and accumulation of P in sediments is closely related to the presence of mineral elements.
Mineral elements also play a role in the release of P in the form of phosphate from sediments into pore water. While Al-bound P and apatite are not actively involved in the release of phosphate from sediments (Gonsiorczyk et al. 1998; Kopáček et al. 2005), it has been reported that Fe- and Mn-bound P in sediments are converted to P sources available to primary producers in the water columns of lakes (Scharf 1999; Zhou et al. 2001). These phenomena are often the main contributors of P to overlying water (Wang et al. 2005) and are believed to cause eutrophication (Correll 1998; Gao et al. 2005). Therefore, the release of phosphate from Fe- and Mn-bound P in sediments plays a significant role in the environmental preservation of lakes, and it is therefore important to examine the mechanisms that control this process.
The release of phosphate from sediments into pore water is influenced by various physical environmental factors, such as temperature, pH, and redox potential (Jensen and Andersen 1992; Hupfer and Lewandowski 2008). These factors vary due to changes in several types of biological activity, such as microorganism community composition, algal blooms, rhizosphere of aquatic plants, and bivalve bioturbation (Törnblom and Rydin 1998; Chen et al. 2016, 2018; Xing et al. 2018). The redox potential in sediments is one of the dominant factors controlling the release of phosphate from Fe- and Mn-bound P (Boström and Pettersson 1982; Gächter et al. 1988; Boström et al. 1989; Smith 2003). The redox potential influences the solubility of Fe and Mn in sediments. Mn-bound P is dissolved and phosphate is released as a consequence in the layers below the Mn redox boundary layer, where the dissolved oxygen content of the sediments is zero (Kawashima et al. 1986; Shaw et al. 1990; Reimers et al. 1992; Rydin 2000). In the same way, Fe-bound P is released as phosphate into the pore water of sediments in the Fe redox boundary layer under conditions where ferric oxides are reduced to ferrous ions (Gomez et al. 1999; Rydin 2000). Recent studies using high-resolution dialysis and diffusive gradients in thin film analyses have verified that the release of P is controlled by Fe redox cycling in sediments (Ding et al. 2016, 2018). Therefore, it is important to understand the changes in the solubilities of Fe and Mn in sediments. The changes in these solubilities are greatly affected by depth-associated decreases in the redox potential. The vertical distribution of sulfur (S) also needs to be considered because the extent of sulfate reduction and sulfide formation increases with depth-associated decreases in the redox potential (Stumm and Morgan 1996).
Lake Kasumigaura is a highly eutrophic lake. The average total P concentration in the lake is 75 μg L−1, and the total mass of P in the water column is 49 metric tons (Havens et al. 2001). Several studies (e.g., Hosomi et al. 1982; Ishii et al. 2010) have demonstrated via incubation experiments that phosphate from the sediments is released into the water column in Lake Kasumigaura. P release in sediments is linked to the vertical distributions of the mineral elements in the sediments. Therefore, observing the vertical and temporal distributions of the mineral elements in the sediments can be helpful to understand the deposition and release of P in sediments. However, few studies have monitored the vertical profiles of these elements in lake sediments in the long term.
In this study, vertical and temporal variations in P, Mn, S, Al, Mg, Ca, and Fe in the sediments of a representative shallow eutrophic lake were investigated using preserved samples. The vertical and temporal profiles of phosphate in the pore water were also analyzed to assess the release of phosphate from the sediments. Based on the data obtained, this study discusses the relationship between the dynamics of these mineral elements and the deposition and release of phosphate from the sediments.
2 Experimental
2.1 Sampling site
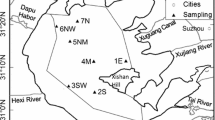
Lake Kasumigaura is the second largest lake in Japan and is a typical eutrophic lake. Lake Kasumigaura has a surface area of 171 km2 and a watershed of approximately 2200 km2. Its mean and maximum depths are 4 m and 7.3 m, respectively. The lake water is used as drinking water and for agricultural and industrial purposes. The watershed is subject to severe anthropogenic impacts because more than one million people live within the watershed and approximately 50% of this land is currently being used for agriculture, either as paddy fields or for other agricultural activities (Fukushima et al. 2007). The center of Lake Kasumigaura (Fig. 1) is the representative monitoring site and is the trend station for the United Nation Global Environment Monitoring System (GEMS)/Water Program. The GEMS/Water trend station is used to monitor long-term changes in water quality associated with a variety of pollution sources and land-use activities. In addition, the Japanese National Institute for Environmental Studies (NIES) has been collecting physical and chemical water quality data on a monthly basis since 1977.

Location of sampling site in Lake Kasumigaura, Japan
2.2 Sample collection
Monthly sediment core sampling was performed from January 8, 2003, to December 10, 2008, in the center of Lake Kasumigaura (140° 24′ 25″ E 36° 01′ 57″ N), which experiences a relatively lower influence from riverine particle inputs than does the lakeshore. Eight sediment cores were collected with an acrylic tube sediment core sampler (30-cm long, 4-cm inner diameter; Rigosha). At each sampling event, eight sediment cores longer than 20 cm were collected. The sediment cores were brought back to our laboratory at the NIES Lake Kasumigaura Water Research Station, located near the lake shore of Lake Kasumigaura, and were sectioned under an N2-purged environment at the following depth intervals: 0–1, 1–2, 2–4, 4–6, 6–8, 8–10, and 10–15 cm. Eight sediment samples from each depth interval were combined as composite samples to reduce the non-uniformity in the sediment cores. The combined samples from the eight cores were placed in a centrifuge tube (500 mL) under an N2-purged environment and then separated into pore water and sediments via centrifugation (Avanti HP-25; Beckman Coulter) at 2278×g for 15 min at 4 °C. The pore water was filtered through a glass fiber filter (nominal pore size 0.7 μm; Whatman) and stored in a polycarbonate bottle at 4 °C or frozen at − 30 °C until further analysis. Sediment samples were freeze-dried, homogenized using a mortar, and stored in a sealed polycarbonate jar until further analysis.
2.3 Elemental analysis
The sediment sample was acid-digested for an elemental analysis via inductively coupled plasma atomic emission spectroscopy (ICP-AES) according to Takamatsu et al. (2010). A 20-mg sediment sample was placed in a 7-mL Teflon vial, and 1 mL of 60% nitric acid (HNO3) (Kanto Chemical) and 1 mL of 60% perchloric acid (Kanto Chemical) were added. The vial was capped and placed in a stainless steel high-pressure digestion bomb. The bomb was heated in an electric oven at 140 °C for 6 h. After cooling, the vial was uncapped and heated on a hot plate at 200 °C with the addition of 0.5 mL of 49.5% hydrofluoric acid (HF) (Kanto Chemical) for approximately 20 min until the sample had evaporated down to a volume of 500 μL. Then, 0.5 mL of 49.5% HF (Kanto Chemical) was again added to the sample to digest the silicate minerals contained within it. The acid-digested sample was diluted to 6 mL with 1% HNO3 and was subjected to an elemental analysis via ICP-AES (ICAP-750; Nippon Jarrell-Ash). Spectral interference caused by high concentrations of Al and Fe in the samples used for the ICP-AES analysis was corrected using the k-factor method (Hee et al. 1985). Acid digestion was performed in duplicate for each sediment sample. The Al, Ca, Fe, Mg, Mn, P, and S contents were measured. The mean ratio of the low to high elemental content in the duplicate samples was 0.97. Analytical precision was verified using certified standard materials designated as Pond Sediment (NIES CRM No.2) (Okamoto and Fuwa 1985) and Lake Sediment (NMIJ CRM 7303a) (Inagaki et al. 2004). The relative standard deviations of five repeat analyses of the certified standard material, Pond Sediment, were less than 1.9% for all elements. The detection limit of ICP-AES was equal to three times the standard deviation of the blank (ultrapure water), and therefore, the limits of Al, Ca, Fe, Mg, Mn, P, and S detection were 0.56 μmol L−1, 0.22 μmol L−1, 0.11 μmol L−1, 0.62 μmol L−1, 0.05 μmol L−1, 1.9 μmol L−1, and 1.9 μmol L−1, respectively.
2.4 Phosphate measurements in pore water
Phosphate concentrations were determined via the molybdenum blue method with an auto analyzer (AACS-II; Bran+Luebbe). The detection limit of phosphate, which was equal to three times the standard deviation of the blank (ultrapure water), was 0.07 μmol L−1.
2.5 Multiple regression analysis
A multiple regression analysis was conducted by setting P as the response variable and the other variables as explanatory variables using the MASS package (Venables and Ripley 2002) in the R environment. The analysis was conducted with standardized variables so that their variances were 1. Several methods are available to determine the optimal subset of response variables, including the Akaike information criterion (AIC) (Akaike 1974) and the Bayesian information criterion (BIC) (Schwarz 1978). Model selection based on AIC has an advantage in exploratory data analyses (Aho et al. 2014). In this study, the model with the minimum AIC score was selected as the optimal model. The variance inflation factor (VIF) was calculated based on the results to evaluate the multicollinearity of the multiple regression model, and it was confirmed that the VIF was less than 10.
3 Results
3.1 Contents of the mineral elements (Fe, Al, Ca, Mg, P, Mn, and S) in the sediment
No large vertical changes were observed in the Fe, Al, Ca, and Mg contents during the study period (Fig. 2), even though the content of these elements exhibited a temporal variation.

Time-depth contours of a iron (Fe), b aluminum (Al), c calcium (Ca), and d magnesium (Mg) contents in Lake Kasumigaura sediment. Data from 0–1, 1–2, 2–4, 4–6, 6–8, 8–10, and 10–15 cm sections are plotted at the 0.5, 1.5, 3, 5, 7, 9, and 12.5 cm depths
No drastic changes in the Fe content were observed from the sediment surface to the bottom layer in 2003 (Fig. 2a). Relatively high values (0.87–0.91 mmol g−1) were observed in the 10–15-cm-depth layer in the summer of 2004 (Table S1, Electronic Supplementary Material). During and after 2006, the Fe content tended to increase in all depth layers compared with those from 2003 to 2005. This trend continued until the end of 2008.
Al was the most abundant of the mineral elements studied and did not show large vertical variations (Fig. 2b). A distinctive temporal variation in the Al content was observed in both 2004 and 2006. The Al content was relatively low from January to June in 2004. A maximum value of 3.4 mmol g−1 was recorded in the 1–2-cm-depth layer in April 2006 (Table S2, Electronic Supplementary Material); after that, the Al content from April 2006 to December 2008 remained higher than the Al content prior to April 2006.
The Ca content showed a temporal variation similar to that of Al (Fig. 2c). From 2003 to 2005, the Ca content tended to decrease in the three lower depth layers (6–15 cm). Conversely, in 2006, the Ca content increased in all depth layers, especially in the four upper depth layers (0–8 cm). The highest Ca content (0.24 mmol g−1) was recorded in the 0–1-cm-depth layer in April 2006 (Table S3, Electronic Supplementary Material); after that, the Ca content remained consistently high until the end of 2008.
The Mg content showed a slight increasing trend with depth (Fig. 2d). The highest Mg content (0.33 mmol g−1) was recorded in the 0–1-cm-depth layer in September 2003 (Table S4, Electronic Supplementary Material). There was no remarkable temporal change in the Mg content in each layer during the study period.
Conversely, the vertical and temporal changes in the P, Mn, and S content in the sediment were sharp (Fig. 3). A linear decrease in the P content ranging from 19 to 45 μmol g−1 was observed from the sediment surface to the bottom layer during the study period (Fig. 3a; Table S5, Electronic Supplementary Material). The P content in the surface layer showed an upward trend after 2005. In fact, the P content was often more than 40 μmol g−1 in the upper 1-cm-depth layer after 2005 (Table S5, Electronic Supplementary Material).

Time-depth contours of a phosphorus (P), b manganese (Mn), and c sulfur (S) contents in Lake Kasumigaura sediment. Data from 0–1, 1–2, 2–4, 4–6, 6–8, 8–10, and 10–15 cm sections are plotted at the 0.5, 1.5, 3, 5, 7, 9, and 12.5 cm depths
The Mn content also exhibited a linear decrease from the sediment surface to the bottom layer in the range of 12–26 μmol g−1 during the study period (Fig. 3b; Table S6, Electronic Supplementary Material). This trend in the Mn content was comparable to that in the P content. The Mn content in the surface layer, especially in the upper 1-cm-depth layer, also showed an increasing trend after 2005.
Throughout the study period, the S content tended to be maximal in the 4–10-cm-depth layer (Fig. 3c). The annual mean S content was 0.17 mmol g−1 in the 6–8-cm-depth layer in 2003 (Table S7, Electronic Supplementary Material). An increasing trend in the S content was observed in the middle layer of the sediments until July 2006. In particular, the S content was often more than 0.20 μmol g−1 in the 4–8-cm-depth layer from May 2005 to July 2006 (Table S7, Electronic Supplementary Material). However, after July 2006, the highest annual mean S content tended to develop in a deeper layer, i.e., the 8–10-cm-depth layer.
3.2 Phosphate concentration in the sediment pore water
The phosphate concentration in the pore water tended to be low in the sediment surface layer and increased in the 2–6-cm-depth layer, particularly in the summer (Fig. 4). Exceptionally, relatively high phosphate concentrations were detected in the lower 2-cm-depth layer in the winter of 2004. By contrast, the pore water phosphate concentration in the middle layer was often lower than 5 μmol L−1 from January to May in 2006. In addition, the phosphate concentration in the pore water in 2008 remained lower than that in the preceding 5 years and exceeded 20 μmol L−1 only once, in the upper 1-cm-depth layer in September (Table S8, Electronic Supplementary Material).

Time-depth contours of phosphate (PO43−) concentrations in Lake Kasumigaura sediment pore water. Data from 0–1, 1–2, 2–4, 4–6, 6–8, 8–10, and 10–15 cm sections are plotted at the 0.5, 1.5, 3, 5, 7, 9, and 12.5 cm depths
3.3 Regression analysis
A multiple linear regression analysis of the P content in the upper 2-cm-depth layer of the sediments of Lake Kasumigaura shows that the P content is significantly related to the Al, Fe, and S contents. The results of this analysis are summarized in Table 1.
4 Discussion
4.1 P accumulation on sediment surfaces
A regression analysis was performed to confirm the presence or absence of the co-precipitation of P with Fe and Al oxide onto the lake sediment. No significant correlation was detected under the 2–4-cm-depth layer. Because this reaction only occurs in the surface layer, the result of the multiple regression analysis exhibited upper 2-cm-depth layer. The results of a multiple regression analysis demonstrated positive correlations between the P, Fe, and Al contents within the 0–2-cm-depth layer (Table 1), suggesting the co-precipitation and accumulation of phosphate with Fe and Al compounds on sediment surfaces (Nair et al. 1995; Andrieux and Aminot 1997). A ferric compound, such as amorphous Fe oxy-hydroxide, can absorb P by forming an Fe(OOH)-P complex (Peng et al. 2007; Wang et al. 2015). In addition, amorphous Al oxides have a high affinity for phosphate and contribute to P binding and retention in sediment (Dapeng et al. 2011; Wang et al. 2015). These findings support our interpretation of the result of the multiple regression analysis for P.
The contributions of other sources for the P accumulation were also considered. The P content in the sediments decreased linearly with depth during the study period (Fig. 3a), and this pattern was similar to that for Mn. Kawashima et al. (1986) reported that Mn oxides adsorb phosphate in the presence of alkali earth metals at a pH of 6–9. This environmental condition is met in the sediments of Lake Kasumigaura (Lake Kasumigaura Database 2018). However, the Mn:P ratio of the sediments (1:2) is too large to be considered as being the adsorption of phosphate to Mn oxide. Therefore, it is unlikely that the coordination of the vertical distributions of Mn and P in the sediments was due to the sedimentation process of the adsorption of phosphate to Mn oxide. There should be only a small contribution from Mg-bound P and Ca-bound P to the accumulation of P in the sediments. This is because the Mg and Ca contents were lower than one third to one tenth of the contents of Fe and Al and no correlation was found between P and Mg and/or Ca in the multiple regression analysis. Even though we did not measure the organic P in the sediment, previous studies have revealed that organic P accounts for less than one third of the total P in the sediments at the center of Lake Kasumigaura (Komatsu et al. 2009; Ishii et al. 2010). Therefore, the contribution of organic P to the P dynamics in the sediment cannot be completely negligible; however, inorganic P will have a greater influence on the total P dynamics than will organic P.
The results of the multiple regression analysis conclusively indicate that P accumulates as Fe-bound P and/or Al-bound P in the surface layer of the Lake Kasumigaura sediments. These findings suggest that elemental analyses of sediments are useful to estimate P accumulation in lake sediments. Other novel approaches (e.g., sequential selective dissolution) or measurements of environmental parameters (e.g., pH, organic carbon content, and carbonate content) should also be helpful to estimate phosphorus dynamics in sediments. However, the approach in our study is likely suitable to investigate the relationship between P and other elements with the appropriate statistical analyses. Our results demonstrate a possible method to examine past lake environments using long-term storable resources such as lake sediments.
4.2 Estimation of the phosphate release layer in sediments based on the Mn and S content
Of the mineral-bound P compounds, Al-bound P and apatite are not actively involved in the release of phosphate from sediments (Gonsiorczyk et al. 1998; Kopáček et al. 2005). Organic P should constitute a phosphate source for the pore water in the sediments of Lake Kasumigaura (Ishii et al. 2010; Shinohara et al. 2012). However, inorganic P accounts for more than two thirds of the total P in the sediments of Lake Kasumigaura (Komatsu et al. 2009; Ishii et al. 2010). Therefore, inorganic P is more likely to become a dominant factor in phosphate release than is organic P. Taking this into consideration, the discussion focuses on the dynamics of inorganic P, especially Fe-bound P, in the sediments.
Ferric oxides in Fe-bound P are reduced to ferrous ions in association with a decrease in the redox potential and the release of phosphate from sediments into pore water (Sundby et al. 1992; Gomez et al. 1999; Rydin 2000). Therefore, an increase in the phosphate concentration in the pore water is generally concomitant with a decrease in the Fe content in the sediments. However, because Fe is abundant in the sediments, the vertical distribution of the Fe content did not show drastic variations and was not substantial enough for the Fe redox boundary layer to be identified (Fig. 2a). Therefore, the location of the Fe redox boundary layer was estimated using the vertical distributions of the more redox-sensitive elements in the sediments.
The Fe redox boundary layer is generally observed between the upper layer of the sulfate reduction and the deeper Mn redox boundary layer (Nealson 1997; Burdige 2006). The Mn redox boundary layer should show the highest Mn oxide content (Burdige 1993). This is because the soluble Mn2+, produced in the Mn redox layer by microbial Mn reduction, is transported in an upward direction and oxidized to insoluble Mn (e.g., MnO2) (Stumm and Morgan 1996). Mn is consequently accumulated in the Mn redox boundary layer; in this study, the Mn content was generally maximized in the 0–1-cm-depth layer of the sediments and decreased with depth (Figs. 3b and 5). In addition, dissolved oxygen in the Lake Kasumigaura pore water was depleted within the 0–1-cm-depth layer of the surface sediments (unpublished data from our laboratory), suggesting that microbial Mn reduction occurs just below this layer (Shaw et al. 1990; Reimers et al. 1992; Pohl et al. 1998). These findings demonstrate that the Mn redox boundary layer generally exists within the 0–1-cm-depth layer in the Lake Kasumigaura sediments.

Depth layer for each observation month in which the maximum phosphate concentration (cross), maximum manganese (dotted line) and sulfur (solid line) contents, and minimum iron content (circle) in Lake Kasumigaura sediment were recorded. Data from 0–1, 1–2, 2–4, 4–6, 6–8, 8–10, and 1–15 cm sections are plotted on the 0.5, 1.5, 3, 5, 7, 9, and 12.5 cm depths
The increase in the S content occurred in the 2–4-cm-depth or deeper sediment layers. Even though there was a temporal variation in the S content, the maximum consistently occurred in the 6–10-cm-depth layer (Figs. 3c and 5). Increases in the S content in sediments are often due to the formation of iron sulfide (FeS) caused by a reaction between dissolved ferrous Fe and sulfide (Golterman 2004; Kraal et al. 2013). This indicates that sulfate reduction and FeS formation occurred actively in the 6–10-cm-depth layer, demonstrating that the sulfate reduction layer likely exists near the 6–10-cm-depth layers of the sediments.
According to the above findings, the Fe redox boundary layer primarily exists near the 1–6-cm-depth layer in the Lake Kasumigaura sediments. Meanwhile, the maximum phosphate concentrations in the pore water often occurred near the 2–6-cm-depth layer of the sediments (Fig. 4), and, therefore, Fe-bound P appeared to be released as phosphate into the sediment pore water in this layer. This layer is comparable to the Fe redox boundary layer, which is estimated using the vertical distributions of the Mn and S contents (Fig. 5). Our results conclusively indicate that the vertical distribution of the sedimentary mineral elements reflects the temporal variation in the redox conditions in the sediment more closely than expected and that it is a useful and effective metric for qualitative assessments of phosphate release from sedimentary P, especially Fe-bound P, to pore water and/or the water column in lake environments.
Our results are likely reliable with respect to estimating the outline of the phosphorus dynamics in sediments; however, the distribution of phosphate in the pore water often changes at the millimeter scale due to the heterogeneity of the sediments. Recent studies have succeeded in observing the millimeter-scale distribution of phosphate and dissolved Fe in sediments using high-resolution methods (Ding et al. 2016; Ding et al. 2018). Therefore, high-resolution methods will be introduced to provide more detailed information about P dynamics in future studies.
5 Conclusions
A multiple regression analysis based on the results of an elemental analysis of the sediments in Lake Kasumigaura suggests that P is co-precipitated with Fe and Al oxides and is deposited onto the sediment surface. The Fe redox boundary layer where phosphate is released from the sediments is generally observed between the Mn redox boundary layer and the sulfate reduction layer, and therefore, the redox boundary layer was estimated based on the vertical distributions of Mn and S. The vertical profiles of Mn and S in the sediments indicated that most Mn ions were oxidized within the 0–1-cm-depth layer and that FeS formation actively occurred in the 6–10-cm-depth layer. Based on these findings, the Fe redox boundary layer is expected to be present near the 1–6-cm-depth layer. This depth layer was found to be consistent with the depth layer at which the maximum pore water phosphate concentrations often occurred. Therefore, it can be concluded that the sediment samples provide important evidence that can disclose the dynamics of P in association with the redox potential in sediments via analyses of the elemental compositions of samples.
References
Aho K, Derryberry DW, Peterson T (2014) Model selection for ecologists: the worldviews of AIC and BIC. Ecology 95:631–636
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Autom Control 19:716–723
Andrieux F, Aminot A (1997) A two-year survey of phosphorus speciation in the sediments of the Bay of Seine (France). Cont Shelf Res 17:1229–1245
Boström B, Pettersson K (1982) Different patterns of phosphorus release from lake sediments in laboratory experiments. Hydrobiologia 91:415–429
Boström B, Pettersson AK, Ahlgren I, Bostrm B (1989) Seasonal dynamics of a cyanobacteria-dominated microbial community in surface sediments of a shallow, eutrophic lake. Aquat Sci 51:153–178
Burdige DJ (1993) The biogeochemistry of manganese and iron reduction in marine sediments. Earth-Science Rev 35:249–284
Burdige DJ (2006) Geochemistry of marine sediments. In: Princeton University Press. Princeton, New Jersey
Chen M, Ding S, Liu L, Xu D, Gong M, Tang H, Zhang C (2016) Kinetics of phosphorus release from sediments and its relationship with iron speciation influenced by the mussel (Corbicula fluminea) bioturbation. Sci Total Environ 542:833–840
Chen M, Ding S, Chen X, Sun Q, Fan X, Lin J, Ren M, Yang L, Zhang C (2018) Mechanisms driving phosphorus release during algal blooms based on hourly changes in iron and phosphorus concentrations in sediments. Water Res 133:153–164
Correll DL (1998) The role of phosphorus in the eutrophication of receiving waters: a review. J Environ Qual 27:261–266
Dapeng L, Yong H, Chengxin F, Yan Y (2011) Contributions of phosphorus on sedimentary phosphorus bioavailability under sediment resuspension conditions. Chem Eng J 168:1049–1054
Ding S, Wang Y, Wang D, Li YY, Gong M, Zhang C (2016) In situ, high-resolution evidence for iron-coupled mobilization of phosphorus in sediments. Sci Rep 6:24341
Ding S, Chen M, Gong M, Fan X, Qin B, Xu H, Gao S, Jin Z, Tsang DCW, Zhang C (2018) Internal phosphorus loading from sediments causes seasonal nitrogen limitation for harmful algal blooms. Sci Total Environ 625:872–884
Fukushima T, Takahashi M, Matsushita B, Okanishi Y (2007) Land use/cover change and its drivers: a case in the watershed of Lake Kasumigaura, Japan. Landsc Ecol Eng 3:21–31
Gächter R, Meyer JS, Mares A (1988) Contribution of bacteria to release and fixation of phosphorus in lake sediments. Limnol Oceanogr 33:1542–1558
Gao L, Zhou JM, Yang H, Chen J (2005) Phosphorus fractions in sediment profiles and their potential contributions to eutrophication in Dianchi Lake. Environ Geol 48:835–844
Goedkoop W, Pettersson K (2000) Seasonal changes in sediment phosphorus forms in relation to sedimentation and benthic bacterial biomass in Lake Erken. Hydrobiologia 431:41–50
Golterman HL (1995) The labyrinth of nutrient cycles and buffers in wetlands: results based on research in the Camargue (southern France). Hydrobiologia 315:39–58
Golterman HL (2004) Sediment and the phosphate cycle. Part 1: speciation, fractionation and bioavailability. In: Golterman HL (ed) The chemistry of phosphate and nitrogen compounds in sediments. Kluwer Academic Publishers, USA, pp 51–96
Gomez E, Durillon C, Rofes G, Picot B (1999) Phosphate adsorption and release from sediments of brackish lagoons: pH, O2 and loading influence. Water Res 33:2437–2447
Gonsiorczyk T, Casper P, Koschel R (1998) Phosphorus-binding forms in the sediment of an oligotrophic and an eutrophic hardwater lake of the Baltic Lake District (Germany). Water Sci Technol 37:51–58
Havens KE, Fukushima T, Xie P, Iwakuma T, James RT, Takamura N, Hanazato T, Yamamoto T (2001) Nutrient dynamics and the eutrophication of shallow lakes Kasumigaura (Japan), Donghu (PR China), and Okeechobee (USA). Environ Pollut 111:263–272
Hee SSQ, Macdonald TJ, Boyle JR (1985) Effects of acid type and concentration on the determination of 34 elements by simultaneous inductively coupled plasma atomic emission spectrometry. Anal Chem 57:1242–1252
Hosomi S, Okada M, Sudo R (1982) Release of phosphorus from lake sediments. Environ Int 7:93–98
Hupfer M, Lewandowski J (2008) Oxygen controls the phosphorus release from lake sediments—a long-lasting paradigm in limnology. Int Rev Hydrobiol 93:415–432
Hupfer M, Gloess S, Grossart HP (2007) Polyphosphate-accumulating microorganisms in aquatic sediments. Aquat Microb Ecol 47:299–311
Inagaki K, Takatsu A, Kuroiwa T, Nakama A, Eyama S, Chiba K, Okamoto K (2004) Certified sediment reference materials for trace element analysis from the National Metrology Institute of Japan (NMIJ). Anal Bioanal Chem 378:1271–1276
Ishii Y, Harigae S, Tanimoto S, Yabe T, Yoshida T, Taki K, Komatsu N, Watanabe K, Negishi M, Tatsumoto H (2010) Spatial variation of phosphorus fractions in bottom sediments and the potential contributions to eutrophication in shallow lakes. Limnology 11:5–16
Jensen HS, Andersen FØ (1992) Importance of temperature, nitrate, and pH for phosphate release from aerobic sediments of four shallow, eutrophic lakes. Limnol Oceanogr 37:577–589
Kawashima M, Tainaka Y, Hori T, Koyama M, Takamatsu T (1986) Phosphate adsorption onto hydrous manganese (IV) oxide in the presence of divalent cations. Water Res 20:471–475
Komatsu N, Ishii Y, Watanabe K, Homma T, Kitamura T, Negishi M, Iwasaki J (2009) Fractionation and distribution of phosphorus as evidence of deposition by fish-farming in eutrophic Lake Kasumigaura, Japan. Jpn J Limnol (Rikusuigaku Zasshi) 69:193–208
Kopáček J, Borovec J, Hejzlar J, Ulrich KU, Norton SA, Amirbahman A (2005) Aluminum control of phosphorus sorption by lake sediments. Environ Sci Technol 39:8784–8789
Kraal P, Burton ED, Bush RT (2013) Iron monosulfide accumulation and pyrite formation in eutrophic estuarine sediments. Geochim Cosmochim Acta 122:75–88
Lake kasumigaura database (2018) http://db.cger.nies.go.jp/gem/moni-e/inter/GEMS/database/kasumi/index.html. Accessed 2 Apr 2018
Nair D, Graetz DA, Portier KM (1995) Forms of phosphorus in soil profiles from dairies of South Florida. Hydrobiologia 59:1244–1249
Nealson KH (1997) Sediment bacteria: who’s there, what are they doing, and what’s new? Annu Rev Earth Planet Sci 25:403–434
Okamoto K, Fuwa K (1985) Certified reference material program at National Institute for Environmental Studies. Anal Sci 1:206–207
Olila OG, Reddy KR, Harris WG (1995) Forms and distribution of inorganic phosphorus in sediments of two shallow eutrophic lakes in Florida. Hydrobiologia 302:147–161
Peng JF, Wang BZ, Song YH, Yuan P, Liu Z (2007) Adsorption and release of phosphorus in the surface sediment of a wastewater stabilization pond. Ecol Eng 31:92–97
Pohl C, Hennings U, Petersohn I, Siegel H (1998) Trace metal budget, transport, modification and sink in the transition area between the Oder and Peene rivers and the southern Pomeranian Bight. Mar Pollut Bull 36:598–616
Reimers CE, Jahnke RA, McCorkle DC (1992) Carbon fluxes and burial rates over the continental slope and rise off Central California with implications for the global carbon cycle. Glob Biogeochem Cycles 6:199–224
Ruttenberg KC (1992) Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnol Oceanogr 37:1460–1482
Ruttenberg KC (2003) The global phosphorus cycle. In: Holland HD, Turekian KK (eds) Treatise on geochemistry, 2nd edn. Elsevier, Amsterdam, pp 499–558
Rydin E (2000) Potentially mobile phosphorus in Lake Erken sediment. Water Res 34:2037–2042
Scharf W (1999) Restoration of the highly eutrophic Lingese Reservoir. Hydrobiologia 416:85–96
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Shaw TJ, Gieskes JM, Jahnke RA (1990) Early diagenesis in differing depositional environments: the response of transition metals in pore water. Geochim Cosmochim Acta 54:1233–1246
Shinohara R, Imai A, Kawasaki N, Komatsu K, Kohzu A, Miura S, Sano T, Satou T, Tomioka N (2012) Biogenic phosphorus compounds in sediment and suspended particles in a shallow eutrophic lake: a 31P-nuclear magnetic resonance (31P NMR) study. Environ Sci Technol 46:10572–10578
Smith VH (2003) Eutrophication of freshwater and coastal marine ecosystems: a global problem. Environ Sci Pollut Res Int 10:126–139
Søndergaard M, Jensen JP, Jeppesen E (2003) Role of sediment and internal loading of phosphorus in shallow lakes. Hydrobiologia 506–509:135–145
Stumm W, Morgan JJ (1996) Oxidation and reduction; equilibria and microbial mediation. In: Stumm W, Morgan JJ (eds) Aquatic chemistry: chemical equilibria and rates in natural waters, 3rd edn. John Wiley & Sons, New Jersey, pp 425–515
Sundby B, Gobeil C, Silverberg N, Mucci A (1992) The phosphorus cycle in coastal marine sediments. Limnol Oceanogr 37:1129–1145
Takamatsu T, Watanabe M, Koshikawa MK, Murata T, Yamamura S, Hayashi S (2010) Pollution of montane soil with Cu, Zn, As, Sb, Pb, and nitrate in Kanto, Japan. Sci Total Environ 408:1932–1942
Törnblom E, Rydin E (1998) Bacterial and phosphorus dynamics in profundal Lake Erken sediments following the deposition of diatoms: a laboratory study. Hydrobiologia 364:55–63
Venables WN, Ripley BD (2002) Linear statistical models. In: Venables WN, Ripley BD (eds) Modern applied statistics with S, 4th edn. Springer, New York, pp 139–182
Wang S, Jin X, Pang Y, Zhao H, Zhou X, Wu F (2005) Phosphorus fractions and phosphate sorption characteristics in relation to the sediment compositions of shallow lakes in the middle and lower reaches of Yangtze River region, China. J Colloid Interface Sci 289:339–346
Wang Y, Zhang M, Zhang D, Shen Z (2015) The influence of sediment particle size on the properties of adsorbed dissolved organic matter in the Yangtze estuary and its interactions with As/Sb. Mar Pollut Bull 105:351–358
Welch EB, Cooke GD (1995) Internal phosphorus loading in shallow lakes: importance and control. Lake Reserv Manag 11:273–281
Xing X, Ding S, Liu L, Chen M, Yan W, Zhao L, Zhang C (2018) Direct evidence for the enhanced acquisition of phosphorus in the rhizosphere of aquatic plants: a case study on Vallisneria natans. Sci Total Environ 616–617:386–396
Zhou Q, Gibson CE, Zhu Y (2001) Evaluation of phosphorus bioavailability in sediments of three contrasting lakes in China and the UK. Chemosphere 42:221–225
Acknowledgments
We thank Drs. M. Nishikawa and T. Sano for supervising the ICP-AES analysis.
Funding
This work was supported by the Environment Research and Technology Development Fund of the Ministry of the Environment, Japan (5-1304). Sampling was supported by the GEMS/Water Trend Monitoring Project at Lake Kasumigaura.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible editor: Shiming Ding
Electronic supplementary material
ESM 1
(DOCX 51 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shimotori, K., Imai, A., Kohzu, A. et al. Long-term observations of the vertical distributions of mineral elements and phosphorus dynamics in sediments in a shallow eutrophic lake in Japan. J Soils Sediments 19, 1566–1575 (2019). https://doi.org/10.1007/s11368-018-2159-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-018-2159-0