
Abstract
The dynamic nature of the mitochondrial network is regulated by mitochondrial fission and fusion, allowing for re-organization of mitochondria to adapt to the cell’s ever-changing needs. As organisms age, mitochondrial fission and fusion become dysregulated and mitochondrial networks become increasingly fragmented. Modulation of mitochondrial dynamics has been shown to affect longevity in fungi, yeast, Drosophila and C. elegans. Disruption of the mitochondrial fission gene drp-1 drastically increases the already long lifespan of daf-2 insulin/IGF-1 signaling (IIS) mutants. In this work, we determined the conditions required for drp-1 disruption to extend daf-2 longevity and explored the molecular mechanisms involved. We found that knockdown of drp-1 during development is sufficient to extend daf-2 lifespan, while tissue-specific knockdown of drp-1 in neurons, intestine or muscle failed to increase daf-2 longevity. Disruption of other genes involved in mitochondrial fission also increased daf-2 lifespan as did treatment with RNA interference clones that decrease mitochondrial fragmentation. In exploring potential mechanisms involved, we found that deletion of drp-1 increases resistance to chronic stresses. In addition, we found that disruption of drp-1 increased mitochondrial and peroxisomal connectedness in daf-2 worms, increased oxidative phosphorylation and ATP levels, and increased mitophagy in daf-2 worms, but did not affect their ROS levels, food consumption or mitochondrial membrane potential. Disruption of mitophagy through RNA interference targeting pink-1 decreased the lifespan of daf-2;drp-1 worms suggesting that increased mitophagy contributes to their extended lifespan. Overall, this work defined the conditions under which drp-1 disruption increases daf-2 lifespan and has identified multiple changes in daf-2;drp-1 mutants that may contribute to their lifespan extension.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are important contributors to organismal health. In addition to being the primary producer of cellular energy, mitochondria also play key roles in apoptosis, calcium regulation, redox homeostasis and inter-organelle communication [1]. In aged organisms, mitochondrial function and mitochondrial morphology become dysregulated [2]. Though the mechanisms by which mitochondria influence longevity are not completely understood, defects in mitochondrial function contribute to multiple age-related metabolic [3,4,5,6,7,8,9], cardiovascular [10, 11] and neurodegenerative diseases [12,13,14,15,16,17].
Mitochondria form an interconnected network within the cell where fusion allows individual mitochondrion to join mitochondrial networks while fission allows mitochondrion to separate from each other. Regulation of mitochondrial fission and fusion allows mitochondria to dynamically respond to environmental conditions [18]. Mitochondrial fission facilitates the clearance of dysfunctional mitochondria as well as the generation of new mitochondria [19,20,21]. Mitochondrial fusion facilitates the complementation of mitochondrial components for optimal mitochondrial function [19, 22,23,24].
As organisms age, their mitochondrial networks become increasingly fragmented and lose the ability to switch between fused and fragmented networks [25,26,27]. Neurons from individuals with neurodegeneration have highly fragmented mitochondrial networks [28,29,30,31]. Furthermore, dysregulated expression of fission and fusion proteins occurs in both aging and age-related disease [2, 16, 32]. Notably, increased mitochondrial fusion has been associated with increased longevity. Healthy human centenarians have highly connected mitochondrial networks compared to 27- and 75-year-old individuals [33]. Additionally, in C. elegans, increased mitochondrial fusion is required for the longevity of multiple long-lived mutants [34,35,36] and overexpression of mitochondrial fusion genes is sufficient to extend longevity and enhance resistance to exogenous stressors (Traa et al., [37] in revision for Aging Cell).
In C. elegans, the mammalian Opa1 homolog, EAT-3, fuses the inner mitochondrial membrane and the mammalian mitofusin homolog, FZO-1, fuses the outer mitochondrial membrane [38,39,40]. The protein responsible for the scission of both mitochondrial membranes in C. elegans, is DRP-1, homolog to the mammalian Drp1 [32, 41]. DRP-1 is recruited by FIS-1, FIS-2, MFF-1 and MFF-2 to the mitochondrial constriction site [42,43,44,45,46], where ER tubules wrap around mitochondria to begin mitochondrial membrane constriction [47, 48]. Oligomerization of DRP-1 into a ring structure occurs at the constriction site where DRP-1 hydrolyzes GTP for the energy needed to complete the scission of the inner and outer mitochondrial membrane [49, 50]. In addition to mitochondrial fission, DRP-1 also mediates peroxisomal fission and thus contributes to the regulation of peroxisomal network morphology [51].
Altering mitochondrial dynamics can affect an organism’s health and longevity. Disruption of mitochondrial fission increases lifespan in yeast and fungal models [52, 53]. In C. elegans, deletion of drp-1 has little or no effect on lifespan in wild-type worms [35, 54] despite increasing resistance to specific exogenous stressors [25]. However, disruption of drp-1 has been shown to affect longevity in other backgrounds. We found that disruption of drp-1 increases lifespan and restores motility in a neuronal model of polyglutamine toxicity [55], but decreases lifespan in a model in which the toxic polyglutamine tract is expressed in muscle [56], suggesting that the beneficial effects of drp-1 inhibition may be tissue-specific. Most notably, disruption of drp-1 has been shown to extend the already long lifespan of daf-2 insulin/IGF-1 receptor mutants and age-1 phosphoinositide 3-kinase (PI3K) mutants [57].
The insulin/IGF-1 signaling (IIS) pathway is highly conserved among animals and links nutrient availability to organismal growth, metabolism and longevity [58]. The long lifespan of IIS pathway mutants, including daf-2 and age-1, is dependent on the activation of the FOXO transcription factor DAF-16 [59]. DAF-16 upregulates the expression of pro-survival genes, such as chaperones and antioxidants, in response to stress, low nutrient availability, or when IIS is disrupted [60].
The mechanism by which drp-1 extends daf-2 lifespan is not known. Previous studies have indicated that drp-1 deletion does not extend daf-2 by enhancing DAF-16 activity, as disruption of drp-1 does not increase DAF-16 activity in wild-type animals [25] or in daf-2 mutants [57]. It has also been shown that the interaction between drp-1 and the IIS pathway is not daf-2-specific as disruption of drp-1 also extends the lifespan of age-1 mutants [57]. Mutants of the IIS pathway have increased mitochondrial function, increased mitochondrial ROS production and increased induction of mitophagy, all of which may be affected by disrupting mitochondrial dynamics [61,62,63].
In this work, we define the conditions under which disruption of drp-1 extends daf-2 longevity and identify multiple factors that are altered by the loss of drp-1 in daf-2 mutants that may contribute to lifespan extension. We find that inhibition of drp-1 during development is sufficient to increase daf-2 lifespan but tissue specific inhibition of drp-1 in the neurons, intestine or muscle fails to extend daf-2 longevity. Additionally, decreasing mitochondrial fragmentation without disrupting drp-1 is also sufficient to increase daf-2 lifespan. We find that disruption of drp-1 increases daf-2 resistance to chronic stress, decreases reproduction, increases mitochondrial function, increases mitophagy and enhances peroxisomal connectivity, all of which may contribute to the effect of drp-1 on daf-2 longevity.
Methods
Strains
N2 | WT |
MQ1753 | drp-1(tm1108) IV |
CB1370 | daf-2(e1370) III |
MQ1770 | daf-2(e1370) III;drp-1(tm1108) IV |
JVR122 | bcIs78(myo-3p::mitoGFP (matrix GFP) + pRF4) I |
JVR218 | drp-1(tm1108); bcIs78(myo-3p::mitoGFP (matrix GFP) + pRF4) I |
JVR644 | daf-2(e1370) III;bcIs78(myo-3p::mitoGFP (matrix GFP) + pRF4) I |
JVR645 | daf-2(e1370) III;drp-1(tm1108) IV;bcIs78(myo-3p::mitoGFP (matrix GFP) + pRF4) I |
IR1631 | Ex003[myo-3p::TOMM-20::Rosella] |
JVR607 | drp-1(tm1108) IV;Ex003[myo-3p::TOMM-20::Rosella] |
JVR646 | daf-2(e1370) III;Ex003[myo-3p::TOMM-20::Rosella] |
JVR647 | daf-2(e1370) III;drp-1(tm1108) IV;Ex003[myo-3p::TOMM-20::Rosella] |
JVR648 | daf-2(e1370) III;hjIs8[ges-1p::GFP-PTS1] |
HC196 | sid-1(qt9) V |
AGD855 | sid-1(qt9) V;uthIs237[myo-3p::tomato + myo3-p::sid-1] |
JVR635 | sid-1(qt9) V;aIxIs6[vha-6p::sid-1::sl2::gfp] |
JVR636 | sid-1(qt9) V;sqIs69[rgef-1p::GFP + rgef-1p::sid-1] |
AGD801 | daf-2(e1370) III;sid-1(qt9) |
MAH410 | daf-2(e1370) III;sid-1(qt9) V;aIxIs6[vha-6p::sid-1::sl2::gfp] |
MAH411 | daf-2(e1370) III;sid-1(qt9) V;uthIs237[myo-3p::tomato + myo-3p::sid-1] |
MAH727 | daf-2(e1370) III;sid-1(qt9);sqIs69[rgef-1p::GFP + rgef-1p::sid 1] |
JVR109 | daf-2(e1370) III;fis-1(tm2227) II |
JVR110 | daf-2(e1370) III;fis-2(gk414) X |
JVR111 | daf-2(e1370) III;fzo-1(tm1133) II |
JVR119 | fis-1(tm2227) II;daf-2(e1370) III;fis-2(gk414) X |
JVR376 | daf-2(e1370);mff-1(tm2955) |
JVR377 | daf-2(e1370);mff-2(tm3041) |
JVR378 | daf-2(e1370);mff-1(tm2955);mff-2(tm3041) |
JVR097 | fis-1(tm2227) II |
JVR079 | fis-2(gk414) X |
JVR076 | fzo-1(tm1133) II |
JVR108 | fis-1(tm2227) II; fis-2(gk414) X |
JVR353 | mff-1(tm2955) |
JVR354 | mff-2(tm3041) |
JVR375 | mff-1(tm2955);mff-2(tm3041) |
JVR079 | eat-3(tm1107) |
daf-2(e1370);eat-3(tm1107) (no strain name because sterile) |
Quantitative real-time RT-PCR
To perform quantitative RT-PCR, we first collected worms in M9 buffer and extracted RNA using Trizol as previously described [64, 65]. Using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems 4368814), the collected mRNA was then converted to cDNA. Quantitative PCR was performed using a PowerUp SYBR Green Master Mix (Applied Biosystems A25742) in a MicroAmp Optical 96-Well Reaction Plate (Applied Biosystems N8010560) and a Viia 7 Applied Biosystems qPCR machine. mRNA levels were calculated as the copy number of the gene of interest relative to the copy number of the endogenous control, act-3, then expressed as a percentage of wild-type. Primer sequences for each target gene are as follows: drp-1 (L- GAGATGTCGCTATTATCGAACG, R- CTTTCGGCACACTATCCTG) dcr-1 (L-ATTTTCGCGTCGTTAGCAGT, R-CGCATCATGTGGAAAATCAC).
Confocal imaging and quantification
Mitochondrial morphology was imaged using worms that express mitochondrially-targeted GFP in the body wall muscle cells. In addition, we utilized a rol-6 mutant background to facilitate imaging of the muscle cells [56]. The rol-6 mutation, which is present on the pRF4 plasmid as a co-injection marker, results in animals moving in a twisting motion, allowing the sheaths of muscle cells to be facing the objective lens and thus more completely facilitates the imaging of mitochondrial networks within cells. Without the rol-6 mutation, only the longitudinal edges of the muscle will often be visible, thus making it difficult to observe mitochondrial organization.
Peroxisomal morphology was imaged using worms that express peroxisome targeted GFP in the intestine. Worms at day 1 or day 8 of adulthood were mounted on 2% agar pads and immobilized using 10 µM levamisole. Worms were imaged under a 40 × objective lens on a Zeiss LSM 780 confocal microscope. Single plane images were collected for a total of twenty-four worms over three biological replicates for each strain. Imaging conditions were kept the same for all replicates and images. Quantification of mitochondrial or peroxisomal morphology was performed using ImageJ. Segmentation analysis was carried out using the SQUASSH (segmentation and quantification of subcellular shapes) plugin. Particle analysis was then used to measure the number, area, circularity, and maximum Feret’s diameter (an indicator of particle length) of the organelles.
The mtRosella mitophagy reporter was imaged as previously described in worms expressing the reporter in the body wall muscle [66]. The whole body of the worm was imaged under a 20 × objective lens on a Zeiss LSM 780 confocal microscope. Quantification of dsRed and GFP fluorescence intensity was performed used ImageJ and representative images show both channels merged.
Thrashing rate
Thrashing rates were determined manually by transferring 20 age-synchronized worms onto an unseeded agar plate. One milliliter of M9 buffer was added and the number of body bends per 30 s was counted for 3 biological replicates of approximately 10 worms per strain.
Brood size
Brood size was determined by placing individual pre-fertile young adult animals onto NGM plates. Worms were transferred to fresh NGM plates daily until progeny production ceased. The resulting progeny was allowed to develop to the L4 stage before quantification. Three biological replicates of 5 animals each were completed.
Oxygen consumption rate
Oxygen consumption measurements were taken using a Seahorse XFe96 analyzer [25, 67]. The night before the assay, probes were hydrated in 200 μL Seahorse calibrant at 37 degrees while the analyzer’s heater was turned off to allow the machine to cool. Day 1 and day 8 worms were collected in M9 buffer and washed three times before being pipetted into a Seahorse 96 well plate (Agilent Technologies Seahorse Flux Pack 103,793–100). Others have previously determined that using between 5–25 worms per well is optimal [68]. Calibration was performed after 22 μL of FCCP and 24 μL sodium azide was loaded into the drug ports of the sensor cartridge. Measurements began within 30 min of worms being added to the wells. Basal oxygen consumption was measured 5 times before the first drug injection. FCCP-induced oxygen consumption was measured 9 times, then sodium-azide induced oxygen consumption was measured 4 times. Measurements were taken over the course of 2 min and before each measurement the contents of each well were mixed for an additional 2 min. Non-mitochondrial respiration (determined by sodium azide-induced oxygen consumption rate) was subtracted from basal respiration to calculate mitochondrial respiration.
ATP determination
Day 1 and day 8 adult worms were collected, washed 3 times and frozen in 50 μL of M9 buffer using liquid nitrogen. Samples were then immersed in boiling water for 15 min followed by ice for 5 min and finally spun down at 14,800g for 10 min at 4 ℃. Supernatants were diluted tenfold before ATP measurements using a Molecular Probes ATP determination kit (A22066) and TECAN plate reader. Luminescence was normalized to protein content measured using a Pierce BCA protein determination kit.
Heat stress assay
To measure resistance to heat stress, approximately 25 pre-fertile young adult worms were transferred to new NGM plates freshly seeded with OP50 bacteria and were incubated at 37℃. Starting at 12 h, survival was measured every hour for a total of 18 h of incubation. Three biological replicates were completed.
Osmotic stress assay
To measure resistance to osmotic stress, approximately 25 pre-fertile young adult worms were transferred to NGM plates containing 700 mM NaCl and seeded with OP50 bacteria. Worms were kept at 20℃ for 24 h before survival was scored. Five biological replicates were completed.
Oxidative stress assays
Resistance to acute oxidative stress was measured by transferring approximately 25 pre-fertile young adult worms to 420 μM juglone plates seeded with OP50 bacteria. Worms were kept at 20℃ and survival was monitored every 2 h for a total of 8 h. Resistance to chronic oxidative stress was performed by transferring 30 pre-fertile young adult worms to freshly prepared plates containing 4 mM paraquat, 100 μM FUdR and seeded with concentrated OP50. Survival was monitored daily. Three biological replicates were completed for both assays.
Anoxic stress assay
To measure resistance to anoxic stress, approximately 50 pre-fertile young adult worms were transferred to new NGM plates seeded with OP50 bacteria. To create a low-oxygen environment for the worms, we utilized Becton–Dickinson Bio-Bag Type A Environmental Chambers. Plates with young adult worms of each strain were placed in the Bio-Bags for 120 h at 20℃, then removed from the bags and allowed to recover for 24 h at 20℃ before survival was measured. Five biological replicates were completed.
Bacterial pathogen stress assay
We tested for nematode resistance to death by bacterial colonization of the intestine. The slow kill assay was performed as previously described [69, 70]. Cultures of Pseudomonas aeruginosa strain PA14 were grown over night for a total of 16 h and then seeded to the center of NGM agar plates containing 25 μM FUdR. Plates were left on the bench to dry overnight and were then incubated at 37℃ for 24 h. Next the plates were left to adjust to the temperature at which the assay is conducted at 25℃ overnight. To begin the assay, approximately 50 age synchronized L4 worms were transferred to each plate. To monitor survival, deaths were scored twice a day until all worms had died. Three biological replicates were completed.
Quantification of ROS levels
ROS levels were measured using dihydroethidium (DHE; ThermoFisher Scientific, D1168), as previously described [71, 72]. A 30 mM solution of DHE in DMSO was aliquoted and stored at − 80 °C. When needed, 5 μl of DHE stock was diluted in 5 mL of PBS to create a 30 μM DHE solution. Age matched day 1 adult worms (approximately 50) were collected in PBS, transferred to a 1.5 mL centrifuge tube and washed 3 times in 1 mL PBS. To immerse worms in a final concentration of 15 μM DHE, 100 μL of PBS was left in the centrifuge tube after the final wash and 100 μL of 30 μM DHE was added to the tube. Centrifuge tubes were wrapped in tinfoil to protect from light and worms were incubated at room temperature, on a shaker for 1 h, then washed 3 times in PBS. For imaging, worms were mounted on a 2% agarose pad and immobilized with 10 mM levamisole. Worms were imaged with the 20 × objective using a Zeiss LSM 780 confocal microscope. A total of 60 worms were imaged over 3 biological replicates, per genotype. Image J was used to quantify the fluorescence intensity of ethidium-labelled ROS in the whole body of the worm. For each image (one worm per image), mean fluorescence intensity of the whole image and background intensity were measured. For each strain, autofluorescence was sampled from approximately 5–10 untreated worms and mean autofluorescence for the strain was determined. Fluorescence intensity of each worm was calculated by subtracting the image's background fluorescence and the strain's mean autofluorescence from the mean fluorescence of the image.
Tetramethylrhodamine (TMRE) staining
We used the potentiometric fluorescent indicator, TMRE, to measure mitochondrial membrane potential. TMRE was dissolved in DMSO to make a 50 mM stock solution. The TMRE stock solution was then diluted in M9 to make a 5 μM working solution. The working TMRE solution was then pipetted onto NGM plates seeded with OP50 and were left on a nutator for 30 min to allow the TMRE to spread evenly over the agar. Approximately 20 age matched L4 or day 7 worms were transferred to the TMRE plates which were then covered to protect from light and stored at 20 ℃ for 20 h. To de-stain, worms were transferred to regular NGM plates seeded with OP50 and left at 20 ℃ for 4 h. Worms were imaged with the 20 × objective using a Zeiss LSM 780 confocal microscope and ImageJ was used to quantify the fluorescence intensity of TMRE in the whole body of the worm.
Lifespan assay
Lifespan assays were completed at 20℃ and on NGM agar plates that contained FUdR to inhibit the development of progeny and limit internal hatching. We used a low concentration of 25 μM FUdR, which we have previously shown does not affect the longevity of wild-type worms [73]. For each lifespan assay, 40 pre-fertile young adult worms were transferred to 25 μM FUdR plates seeded with OP50 bacteria and were kept at 20℃. Four biological replicates were started on four subsequent days and all replicates were scored every other day to monitor survival until all worms died. Worms were excluded from the assay if they crawled off the agar and died on the side of the plate, had internal hatching of progeny or expulsion of internal organs. Raw lifespan data can be found in Table S1.
RNA interference (RNAi)
We used RNAi to inhibit the expression of specific genes. RNAi clones from the Ahringer library were streaked onto LB plates containing 50 μg/mL carbenicillin and 10 ug/ml Tetracycline. Individual colonies were then inoculated in 2YT media with 50 μg/mL carbenicillin and grown with aeration at 37 ℃ for 18 h. For experiments testing the tissue and timing requirements for drp-1 knockdown to extend lifespan, RNAi clones were sequence verified and were concentrated before being seeded onto NGM plates containing 3 mM IPTG and 50 μg/mL carbenicillin. Seeded RNAi plates were left on the bench for two days to induce dsRNA expression in the bacteria. When conducting lifespan assays, we used the L4 parental paradigm where RNAi knockdown is begun in the parental generation. Age matched L4 worms were transferred to RNAi plates for 24 h after which they were transferred to another RNAi plate for 24 h before being removed. The progeny from these worms were then transferred to the experimental RNAi plates containing 25 μM FUdR for lifespan assays. RNAi lifespan assays were conducted at 20 ℃ where deaths were scored every two days and a minimum of three biological replicates were completed.
Food consumption
The relative amount of growth-arrested OP50 bacteria was quantified before and after incubation with C. elegans in liquid culture, as previously described [74, 75]. Bacterial growth was arrested by resuspending a fresh bacterial pellet in S-basal medium containing 5 mg/L cholesterol, 50 mg/L ampicillin, 10 mg/L kanamycin and 1 mg/L tetracycline. The concentration of the resuspended bacteria was 1.25 OD (600 nm). The bacteria culture was left at room temperature for 7 h and then placed at 4 °C for one week before the assay. On the day of the assay, the S-basal medium was replaced with fresh media containing 100 μM FUdR, maintaining the same bacterial concentration and antibiotic treatment. Age-matched L4 worms were collected with M9 buffer and transferred to a 24-well plate, placing 25–30 worms per well. Then, 1 mL of the medium containing the arrested bacteria was added to each well and the initial OD (600 nm) was measured. Worms were incubated at 20 °C on an orbital shaker at 80 RPM for 4 days. The plate was sealed and placed inside a box containing a small amount of water during the incubation to prevent evaporation. After the incubation period, the OD (600 nm) was measured again and the change in OD for each well was normalized to the number of worms. Three independent assays were performed, with 2 wells per strain in each of the independent assays.
Pharyngeal pumping
To measure pharyngeal pumping 10 pre-fertile young adult worms were transferred to NGM plates seeded with OP50 bacteria. Using the 80 × magnification of a Leica MZ10 F Stereo Microscope, the number of pharyngeal contractions were counted for 2 min.
Statistical analysis
A minimum of three biological replicates were completed for all assays, except where noted. Where possible, the experimenter was blinded to the genotype during the course of the experiment, to ensure unbiased results. Statistical significance of differences between groups was determined by computing a t-test, a one-way ANOVA, a two-way ANOVA or a log-rank test using Graphpad Prism, as indicated in the figure legends. All error bars indicate the standard error of the mean.
Results
Disruption of drp-1 extends lifespan and enhances resistance to chronic stress in daf-2 mutants
To investigate the role of mitochondrial dynamics in the longevity of daf-2 mutants, we disrupted the drp-1 gene in daf-2 mutants and measured their lifespan. While disruption of drp-1 provides a slight but significant extension of lifespan in wild-type worms (Fig. S1), disruption of drp-1 drastically extends the lifespan of already long-lived daf-2 mutants (Fig. 1A). In addition to having a significantly longer lifespan, daf-2 worms have an increased healthspan [76, 77] and increased resistance to stress [78,79,80] but reduced fecundity [81,82,83]. Thus, to evaluate how disruption of drp-1 affects the physiology and biological resilience of daf-2 worms, we measured motility (thrashing rate), fertility (brood size), and resistance to chronic oxidative stress (4 mM paraquat), acute oxidative stress (420 µM juglone), bacterial pathogen stress (P. aeruginosa, PA14), heat stress (37 ℃), osmotic stress (700 mM NaCl), and anoxic stress (72 h, 24 h recovery).

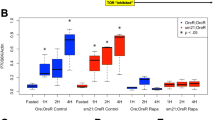
Disruption of drp-1 extends the already long lifespan of daf-2 mutants and increases resistance to bacterial pathogens and chronic oxidative stress. Deletion of drp-1 markedly increases the lifespan of daf-2 mutants (A; daf-2 N = 79, daf-drp-1 N = 165, temperature = 20 °C). Loss of drp-1 reduces the rate of movement (B) and brood size (C) of daf-2 worms. Deletion of drp-1 increases daf-2 resistance to chronic oxidative stress (D; 4 mm paraquat) and bacterial pathogen stress (E; P. aeruginosa strain PA14). In contrast, daf-2;drp-1 mutants have increased sensitivity to heat stress (F) and acute oxidative stress (G). Deletion of drp-1 does not affect resistance to osmotic stress (H) or anoxia (I) in daf-2 worms. A minimum of three biological replicates were performed. Raw lifespan data can be found in Table S1. Statistical significance was assessed using a log-rank test in panels A, D, E and G; a two-way ANOVA with Šidák’s multiple comparisons test for panel B and F; and a student’s t-test for panels C, H, and I. Error bars indicate SEM. **p < 0.01, ****p < 0.0001
Disruption of drp-1 slightly but significantly decreased the thrashing rate of daf-2 animals at day 1, 4 and 8 of adulthood but had no effect at day 12 and 18 (Fig. 1B). Disruption of drp-1 further decreased the brood size of daf-2 worms such that daf-2;drp-1 double mutants produce few viable eggs (Fig. 1C). The drp-1 deletion also slowed the development rate of daf-2 worms (we observed that the time to develop to adulthood is delayed in daf-2;drp-1 worms but did not quantify the magnitude of this delay). Disruption of drp-1 had similar effects in wild-type worms (effect of drp-1 disruption in daf-2 versus wild-type background is summarized in Table S2).
We and others have shown that daf-2 mutants have increased resistance to exogenous stressors [84, 85]. Disruption of drp-1 in daf-2 mutants further increased resistance to chronic stressors including chronic oxidative stress (Fig. 1D; 4 mM paraquat) and bacterial pathogen stress (Fig. 1E; P. aeruginosa strain PA14). In contrast, disruption of drp-1 decreased daf-2 resistance to heat stress (Fig. 1F; 37°C) and acute oxidative stress (Fig. 1G; 420 µM juglone) but had no effect on resistance to osmotic stress (Fig. 1H; 700 mM NaCl) or anoxic stress (Fig. 1I; 120 h). Combined these results show that the drp-1 deletion slows movement, decreases fertility, delays development and enhances resistance to specific stressors in daf-2 worms, all of which have been associated with increased lifespan.
Disruption of drp-1 increases the connectivity of the mitochondrial and peroxisomal networks in daf-2 mutants
Using a strain expressing mitochondrially-targeted GFP in muscle cells, we examined how disruption of drp-1 affects mitochondrial network morphology at day 1 and day 8 of adulthood in daf-2 and wild-type animals. Similarly, to determine if disruption of drp-1 affects peroxisomal network morphology in daf-2 worms, we visualized peroxisomes using a strain expressing peroxisome-targeted GFP in the intestine.
At day 1 of adulthood, disruption of drp-1 produced a hyperfused mitochondrial network morphology in daf-2 mutants, where rather than being organized into parallel elongated tubules, larger tubules and aggregated mitochondria were interconnected by thin filaments (Fig. 2A). Quantification of mitochondrial morphology revealed that deletion of drp-1 in daf-2 worms resulted in a decrease in the number of mitochondria, an increase in the average area of a mitochondrion, an increase in mitochondrial circularity and a decrease in the length of mitochondria (Fig. 2A). At day 8 of adulthood, the mitochondrial network of daf-2;drp-1 mutants continues to appear more connected compared to the tubular morphology of daf-2 mitochondrial networks, but both show an increase in mitochondrial fragmentation compared to day 1 adult worms as indicated by an increase in the number of mitochondria (Fig. 2B). Disruption of drp-1 also increased mitochondrial connectivity in wild-type worms at day 1 and day 8 of adulthood (Fig. S2, S3). Our previous work has shown that disruption of the mitochondrial fusion genes fzo-1 or eat-3 has the opposite effect of increasing mitochondrial fragmentation (Table S3) [25].

Disruption of drp-1 increases mitochondrial and peroxisomal connectivity in daf-2 mutants. At day 1 of adulthood, deletion of drp-1 decreases increases mitochondrial connectivity in daf-2 worms. daf-2;drp-1 worms exhibit decreased mitochondrial numbers, increased mitochondrial area, increased mitochondrial circularity and decreased mitochondrial length compared to daf-2 worms (A). At day 8 of adulthood, deletion of drp-1 decreases mitochondrial number while increasing circularity in daf-2 worms (B). Decreasing drp-1 levels with RNAi also increases the connectivity of the peroxisomal network. Quantification of peroxisome morphology indicates that daf-2;drp-1 worms have decreased peroxisome numbers, decreased peroxisome circularity and increased peroxisome length compared to daf-2 worms (C). Three biological replicates were imaged. Statistical significance was assessed using a student’s t-test. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bar indicates 25 µm in panels A and B, and 10 µm in panel C
Visualization of daf-2 intestinal peroxisomes at day 1 of adulthood revealed that RNAi inhibition of drp-1 increased peroxisomal network connectivity (Fig. 2C). In daf-2 animals that were fed drp-1 RNAi, peroxisomal tubules were visible resulting in a decrease in the number of peroxisomes, a decrease in peroxisomal circularity and an increase in peroxisomal length compared to daf-2 animals that were fed empty vector bacteria. Similarly, drp-1 RNAi increased peroxisomal connectivity in wild-type animals (Fig. S4). Thus, disruption of drp-1 increases the connectivity of both the mitochondrial and peroxisomal networks in daf-2 mutants and wild-type worms.
Inhibition of drp-1 during development is sufficient to extend daf-2 lifespan
In order to define the conditions under which drp-1 disruption extends daf-2 lifespan, we determined when during the animal’s life disruption of drp-1 is required to extend daf-2 lifespan. To do this, we fed daf-2 and wild-type animals drp-1 RNAi either during development only, during adulthood only, or during both development and adulthood and we measured their lifespans. Knockdown during adulthood was achieved by growing worms on empty vector bacteria and transferring worms to drp-1 RNAi plates at day 1 of adulthood (Fig. 3A). Development only knockdown of drp-1 was achieved by growing worms on drp-1 RNAi until adulthood and then transferring these worms to RNAi targeting dicer (dcr-1), which is required for RNAi activity, in order to inhibit the knockdown of drp-1 [86,87,88].

Inhibition of drp-1 during development is sufficient to extend daf-2 lifespan. A To determine when drp-1 depletion acts to increase daf-2 lifespan, drp-1 levels were reduced during development only (drp-1 to dcr-1; blue), during adulthood only (EV to drp-1; red) or during development and adulthood (drp-1 to drp-1; purple) and compared to empty vector (EV) from development to adulthood (EV to EV; white bar or black line). To confirm that drp-1 mRNA levels were being affected as anticipated, wild-type and daf-2 worms were treated with RNAi according to the paradigms outlined in panel A and mRNA was measured using quantitative RT-PCR. B Measurement of drp-1 levels by quantitative RT-PCR confirmed that drp-1 levels were decreased during adulthood by drp-1 RNAi. C Worms transferred to dcr-1 RNAi exhibit decreased levels of dcr-1 mRNA. D Worms treated with drp-1 RNAi during development and dcr-1 RNAi during adulthood show a recovery of drp-1 mRNA levels during adulthood. In wild-type worms, adulthood only knockdown of drp-1 did not affect lifespan (E), while development only drp-1 RNAi decreased longevity (F). In daf-2 mutants, adult only drp-1 RNAi did not affect longevity (G), while drp-1 knockdown during development significantly increased lifespan (H). In panels (B), (C), and (D), three biological replicates were performed in wild-type worms and two biological replicates in daf-2 animals. Four biological replicates were performed for panels (E–H). For lifespan experiments, EV to EV N = 167, EV to drp-1 N = 166, drp-1 to drp-1 N = 174, EV to dcr-1 N = 147, drp-1 to dcr-1 N = 181, temperature = 20 °C. Raw lifespan data can be found in Table S1. Statistical significance was assessed using a one-way ANOVA with Dunnett’s multiple comparisons test in panels B, C, and D; and a log-rank test in panels E–H. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Prior to beginning the lifespan experiments, we measured mRNA levels of drp-1 and dcr-1 in wild-type and daf-2 worms to ensure that drp-1 expression was being knocked down as expected. Adult only knockdown of drp-1 significantly decreased the levels of drp-1 mRNA within 1 day of transferring to drp-1 RNAi (Fig. 3B). The level of knockdown achieved was equivalent to the level in worms with life long drp-1 knockdown. For the development only paradigm, we confirmed that transferring to dcr-1 RNAi decreased levels of dcr-1 transcripts (Fig. 3C) and resulted in a recovery in the level of drp-1 transcripts (Fig. 3D) one day after being transferred.
Wild-type animals with adulthood drp-1 knockdown had no change in lifespan compared to those which were fed empty vector bacteria throughout their lives (Fig. 3E). However, wild-type animals with developmental or lifelong drp-1 knockdown had shorter lifespans compared to those that were fed empty vector bacteria (Fig. 3F). In daf-2 mutants, animals with adulthood drp-1 knockdown did not live longer than animals treated with empty vector (Fig. 3G). However, daf-2 mutants with developmental drp-1 knockdown lived significantly longer than animals fed empty vector bacteria, such that their survival curve was equivalent to that of animals with lifelong knockdown of drp-1 (Fig. 3H). Thus, disruption of drp-1 during development is sufficient to increase daf-2 lifespan.
Tissue specific inhibition of drp-1 in the muscle, neurons or intestine is not sufficient to extend daf-2 lifespan
To determine the extent to which inhibition of drp-1 in specific tissues is sufficient to extend daf-2 lifespan, we used RNAi to knockdown drp-1 expression in the neurons, muscle and intestine of daf-2 and wild-type animals. To achieve a tissue-specific knockdown of drp-1, we used sid-1 mutants, which lack the dsRNA importer SID-1 in all of their tissues, and re-expressed sid-1 from tissue specific promoters, resulting in tissue-specific RNAi sensitivity. The tissue-specific promoters that we used were vha-6p for the intestine, myo-3p for the muscle and rgef-1p for the neurons.
In order to confirm the tissue-specific RNAi sensitivity of each strain, we knocked down genes that act in each specific tissue to produce a clear phenotype. We knocked down the intestinal elt-2 gene, which causes L1 arrest (Fig. 4A), the hypodermal bli-3 gene, which causes cuticle blistering (Fig. 4B), the muscular pat-4 gene, which causes paralysis (Fig. 4C), and the neuronal unc-70 gene, which causes uncoordinated movement (Fig. 4D). daf-2 worms were sensitive to all four RNAi treatments, while daf-2;sid-1 animals were completely resistant. We found that animals re-expressing sid-1 from intestine-specific vha-6p were only sensitive to elt-2 RNAi and animals re-expressing sid-1 from the muscle-specific myo-3p were only sensitive to pat-4 RNAi. Animals re-expressing sid-1 from neuronal-specific rgef-1p were sensitive to unc-70 RNAi and slightly sensitive to RNAi in the intestine. Combined, this indicates that the tissue-specific RNAi strains were functioning as anticipated to limit RNAi knockdown to the targeted tissue.

Inhibition of drp-1 in individual tissues fails to extend daf-2 lifespan. To identify the tissues in which decreasing the levels of drp-1 acts to extend daf-2 lifespan, tissue-specific RNAi was used to lower drp-1 levels in different tissues of daf-2 mutants and wild-type controls. To confirm that tissue-specific RNAi was working, all of the tissue-specific RNAi strains were treated with RNAi that should only produce a phenotype when knocked down in intestine (A; elt-2 RNAi leading to arrestment and B; bli-3 RNAi leading to blistering), body wall muscle (C; pat-4 RNAi leading to paralysis) or neurons (D; unc-70 RNAi leading to uncoordinated movement). In each case, treatment with RNAi caused a phenotype in daf-2 worms and the corresponding tissue-specific RNAi strain but not daf-2;sid-1 worms or any of the other untargeted tissue-specific RNAi strains, thereby indicating that the strains exhibit tissue specificity. In a wild-type background, knocking down drp-1 in the intestine had no effect on lifespan (E), while knocking down drp-1 in muscle (F) or neurons (G) resulted in a small increase in lifespan. In daf-2 worms, knocking down drp-1 in the intestine (H) body wall muscle (I) or neurons (J) had no effect on longevity. Three biological replicates were performed in panels A-D and four biological replicates were performed in panels E-J. For lifespan experiments, sid-1 + drp-1 RNAi N = 185, Intestine + drp-1 RNAi N = 186, Muscles + drp-1 RNAi N = 178, Neurons + drp-1 RNAi N = 207, daf-2;sid-1 + drp-1 RNAi N = 143, daf-2;Intestines + drp-1 RNAi N = 133, daf-2;Muscles + drp-1 RNAi N = 138, daf-2;Neurons + drp-1 RNAi N = 150, temperature = 20 °C. Raw lifespan data can be found in Table S1. Intestine = re-expression of sid-1 in the intestine, Muscles = re-expression of sid-1 in the body wall muscles, Neurons = re-expression of sid-1 in neurons. Statistical significance was assessed using a one-way ANOVA with Dunnett’s multiple comparisons test in panels A-D and a log-rank test in panels E-L. In panels A-D, statistically significant differences from daf-2;sid-1 are shown. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. A comparison of drp-1 RNAi to empty vector can be found in Fig. S5
In considering whether inhibition of drp-1 in a specific tissue is sufficient to extend lifespan, we compared the survival curve of animals with tissue-specific drp-1 knockdown to two controls: (1) animals with tissue-specific RNAi sensitivity that were grown on empty vector bacteria; and (2) sid-1 mutants, which have whole-body resistance to RNAi. We considered a tissue-specific knockdown of drp-1 to affect lifespan if its curve was statistically different from that of its empty vector control as well as the sid-1 mutant control.
In wild-type animals, inhibition of drp-1 in the intestine increased lifespan compared to the empty vector control (Fig. S5A) but not compared to sid-1 mutants (Fig. 4E). Inhibition of drp-1 in the muscle of wild-type animals did not affect lifespan compared to the empty vector control (Fig. S5B) but increased lifespan compared to sid-1 mutants (Fig. 4F). Pan-neuronal inhibition of drp-1 in wild-type animals increased lifespan compared to both the empty vector control (Fig. S5C) and sid-1 mutants (Fig. 4G). Combined, this indicates that disruption of drp-1 in neurons promotes longevity.
In daf-2 animals, inhibition of drp-1 in the intestine, muscle or neurons had no effect on lifespan compared to either the empty vector (Fig. S5D-F) or sid-1 control (Fig. 4H-J). Overall, these results demonstrate that drp-1 knockdown in intestine, muscle or neurons is not sufficient to extend daf-2 lifespan.
Disruption of other mitochondrial fission genes increases daf-2 lifespan
To determine the extent to which drp-1’s role in mitochondrial fission contributes to lifespan extension in daf-2;drp-1 double mutants, we evaluated whether disruption of other genes involved in mitochondrial fission could also increase longevity in daf-2 worms. We found that disruption of fis-1 or fis-1 and fis-2 together significantly increases daf-2 lifespan, while deletion of fis-2 alone does not extend longevity (Fig. 5A-C). Similarly, disruption of mff-1 and mff-2 together extends daf-2 lifespan, while single deletions in either gene have no effect (Fig. 5D-F). In contrast, disruption of fis-1, fis-2, mff-1, mff-2 or combinations of these genes do not extend the lifespan of wild-type worms (Fig. S6A-F). Combined, these results demonstrate that disrupting other genes involved in mitochondrial fission can also increase the lifespan of daf-2 worms.

Disruption of mitochondrial fission genes increases daf-2 lifespan. Deletion of the mitochondrial fission gene fis-1 (A) or fis-1 and fis-2 together (C) increases daf-2 lifespan, while disruption of fis-2 results in a small decrease in daf-2 lifespan (B). Disruption of the mitochondrial fission factor genes mff-1 (D) or mff-2 (E) does not affect daf-2 lifespan individually but together results in a significant increase in daf-2 longevity (F). In contrast to the ability of drp-1 deletion to increase daf-2 lifespan, disruption of fzo-1 does not significantly affect daf-2 lifespan (G). Disruption of eat-3 markedly extends daf-2 lifespan but also results in sterility (H). Three biological replicates were performed. For lifespan experiments, daf-2 N = 79, daf-2;fis-1 N = 115, daf-2;fis-2 N = 102, daf-2;fis-1;fis-2 N = 105, daf-2;mff-1 N = 108, daf-2;mff-2 N = 68, daf-2;mff-1;mff-2 N = 95, daf-2;fzo-1 N = 65, daf-2;eat-3 N = 228, temperature = 20 °C. Raw lifespan data can be found in Table S1. Statistical significance was assessed using the log-rank test
Since disruption of mitochondrial fission increases daf-2 lifespan, we next evaluated whether disruption of mitochondrial fusion genes in daf-2 worms would have the opposite effect. We found that disruption of fzo-1 did not affect lifespan in daf-2 mutants (Fig. 5G) or wild-type worms (Fig. S6G). While disruption of eat-3 caused daf-2 mutants to become sterile, the sterile daf-2;eat-3 double mutants lived significantly longer than daf-2 worms (Fig. 5H). Disruption of eat-3 also extends lifespan in wild-type worms (Fig. S6H). Given that disruption of eat-3 has multiple effects in daf-2 worms, it is unclear whether the mechanism underlying lifespan extension is due to eat-3’s role in mitochondrial fusion, the effect of the eat-3-induced sterility on daf-2 lifespan or the effect of dietary restriction on daf-2 longevity (eat mutants have decreased feeding).
Decreasing mitochondrial fragmentation without disrupting mitochondrial fission machinery can extend daf-2 lifespan
As mitochondrial fission is important for the function of the cell, we tested whether decreasing mitochondrial fragmentation, without directly disrupting the mitochondrial fission machinery, could still extend daf-2 lifespan. To do this, we treated daf-2 worms with twenty-six RNAi clones that were previously shown to decrease mitochondrial fragmentation in body wall muscle [89]. Of the twenty-six RNAi clones tested, ten RNAi clones significantly extended the lifespan of daf-2 mutants (Fig. 6A), one decreased the lifespan of daf-2 mutants and fifteen did not affect lifespan. RNAi clones that extended daf-2 lifespan include sdha-2 (Fig. 6B), C34B2.8 (Fig. 6C), K02F3.2 (Fig. 6D), T10F2.2 (Fig. 6E), Y69F12A.b (Fig. 6F), Y69F12A.c (Fig. 6G), timm-17B.1 (Fig. 6H), C33A12.1 (Fig. 6I), cyp-35A1 (Fig. 6J) and pgp-3 (Fig. 6K). These genes are involved in various pathways of metabolism, protein transport or oxidative phosphorylation and are not characterized as directly contributing to mitochondrial fission or fusion processes [89]. We previously found that some of these RNAi clones increase lifespan in wild-type worms (Table S4) and models of polyglutamine toxicity [55], while others do not.

Decreasing mitochondrial fragmentation without disrupting drp-1 can extend daf-2 lifespan. To determine if decreasing mitochondrial fragmentation independently of drp-1 could extend daf-2 lifespan, daf-2 worms were individually treated with 26 RNAi clones that were previously shown to decrease mitochondrial fragmentation in wild-type worms. Of these 26 RNAi clones, ten RNAi clones significantly increased the lifespan of daf-2 mutants. This suggests that decreasing mitochondrial fragmentation may be sufficient to extend longevity in daf-2 worms. RNAi clones that increased lifespan compared to control (blue) are shown in green. None of these clones decrease the levels of drp-1 mRNA (Fig. S7).Three biological replicates were performed. For lifespan experiments, the sample size is indicated above each bar in panel A. Lifespan was performed at 20 °C. Raw lifespan data can be found in Table S1. Statistical significance was assessed using the log-rank test. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
To determine if these genes are increasing daf-2 lifespan by decreasing drp-1 levels, we measured the effect of the 10 lifespan-extending RNAi clones on drp-1 mRNA levels using quantitative RT-PCR. We found that none of the 10 RNAi clones decreased the levels of drp-1 mRNA (Fig. S7) indicating that their effect on daf-2 lifespan is not through decreasing drp-1 levels. Combined, these results show that decreasing mitochondrial fragmentation, either through disruption of other mitochondrial fission genes (fis-1, fis-2, mff-2, mff-2) or genes that affect mitochondrial fragmentation but are not directly involved in mitochondrial fission, can be sufficient to increase daf-2 lifespan without disrupting drp-1.
Disruption of drp-1 does not affect levels of ROS in daf-2 mutants
daf-2 worms and a number of other long-lived mutants have been shown to have increased levels of ROS, which contributes to their lifespan extension [63, 90, 91]. As mitochondria are the primary site of ROS production in the cell, we asked whether disruption of drp-1 might be altering ROS levels in daf-2 worms and contributing to the extended lifespan of daf-2;drp-1 double mutants. To determine whether ROS levels are altered by disruption of drp-1 in either wild-type or daf-2 mutants, we measured the fluorescence intensity of worms treated with the ROS-sensitive fluorescent dye dihydroethidium (DHE) at day 1 and day 8 of adulthood. While disruption of drp-1 significantly increased the fluorescence intensity of DHE in wild-type animals, it did not further increase ROS levels in daf-2 mutants (Fig. S8A,B). This suggests that altering ROS levels does not contribute to the effect of drp-1 deletion on daf-2 longevity.
Disruption of drp-1 does not affect mitochondrial membrane potential in daf-2 mutants
Alterations in mitochondrial membrane potential have been associated with longevity. daf-2 mutants and several other long-lived mutants were shown to have lower mitochondrial membrane potential than wild-type worms, though others have reported the opposite [62, 92]. Mitochondrial membrane potential decreases with age [93] and preventing this decline is sufficient to increase lifespan [94]. Moreover, dietary restriction appears to increase lifespan at least partially through the preservation of the mitochondrial membrane potential [95]. Accordingly, we examined the effect of drp-1 disruption on mitochondrial membrane potential in wild-type and daf-2 worms to assess its potential contribution to lifespan extension.
To quantify mitochondrial membrane potential, we imaged day 1 and day 8 worms treated with the TMRE (tetramethylrhodamine ethyl ester), a fluorescent indicator whose uptake is dependant on mitochondrial membrane potential. At both time points, we found that daf-2 worms have decreased mitochondrial membrane potential compared to wild-type animals (Fig. S9A,B). While disruption of drp-1 decreased TMRE fluorescence in wild-type animals, it had no effect on TMRE fluorescence in daf-2 mutants (Fig. S9A,B). This suggests that drp-1 deletion does not increase daf-2 lifespan through altering mitochondrial membrane potential.
Disruption of drp-1 does not affect food consumption in daf-2 mutants
Dietary restriction has been shown to extend lifespan in C. elegans as well as many other organisms [96, 97]. daf-2 worms have been shown to have decreased food consumption [75] and daf-2 worms with either an eat-2 mutation [96, 98] or eat-3 mutation (Fig. 5) have increased lifespan. To determine whether disruption of drp-1 might be increasing daf-2 lifespan by decreasing food consumption, we compared food consumption in daf-2 and daf-2;drp-1 animals by directly measuring the decrease in bacteria concentration over time in liquid (Fig. S10A) and by measuring pharyngeal pumping rates (Fig. S10B). In both cases, we found that daf-2 worms have decreased food consumption compared to wild-type animals but that inhibition of drp-1 did not affect food consumption. Thus, decreased food consumption does not contribute to the effect of drp-1 disruption on daf-2 lifespan.
Disruption of drp-1 increases mitochondrial function during young adulthood in daf-2 mutants
Previous studies have reported alterations in mitochondrial function are associated with increased lifespan [62, 99,100,101,102,103,104,105,106]. To investigate whether disruption of drp-1 affects mitochondrial function in daf-2 worms, we measured oxygen consumption and ATP content at day 1 and day 8 of adulthood.
In day 1 adults, disruption of drp-1 increases the oxygen consumption rate of daf-2 mutants, but not of wild-type animals (Fig. 7A). Furthermore, in day 1 adults, disruption of drp-1 increases ATP levels in daf-2 mutants, but not in wild-type animals (Fig. 7B). In day 8 adults, disruption of drp-1 does not affect the oxygen consumption rate (Fig. 7C) or ATP levels (Fig. 7D) of either wild-type or daf-2 mutants. Combined these results show that disruption of drp-1 further improves the mitochondrial function of daf-2 mutants early in adulthood, which may contribute to their lifespan extension.

Disruption of drp-1 increases mitochondrial function in daf-2 mutants at day 1 of adulthood. While drp-1 mutants consumed similar levels of oxygen as wild-type animals, daf-2;drp-1 mutants consumed more oxygen than daf-2 mutants at day 1 of adulthood (A). At day 8 of adulthood, disruption of drp-1 does not affect oxygen consumption in daf-2 or wild-type worms (B). Similarly, daf-2;drp-1 worms showed increased levels of ATP content compared to daf-2 animals at day 1 of adulthood (C) but no difference at day 8 of adulthood (D). drp-1 mutants showed no change in ATP content compared to wild-type worms at day 1 or day 8 of adulthood. Three biological replicates were performed. Statistical significance was assessed using a two-way ANOVA with Šidák’s multiple comparisons test. Error bars indicate SEM. *p < 0.05, **p < 0.01
Disruption of drp-1 increases levels of mitophagy in daf-2 mutants
Increased induction of mitophagy is observed in daf-2 mutants [61] and has been shown to extend lifespan [107, 108]. As mitochondrial fission can facilitate mitophagy, we assessed whether disruption of drp-1 affects levels of mitophagy in daf-2 mutants. To do this, we generated daf-2 animals expressing the mitochondrial-targeted Rosella (mtRosella) biosensor. mtRosella uses a pH-insensitive red fluorescent protein (dsRed) fused to a pH-sensitive green fluorescent protein (GFP) to monitor mitophagy levels in the body wall muscle cells of C. elegans [66]. After imaging the worms and quantifying the fluorescence intensity emitted by each protein, the dsRed to GFP ratio indicates levels of mitophagy. As expected, mitophagy levels were increased in daf-2 mutants, compared to wild-type animals. Disruption of drp-1 induces increased levels of mitophagy in daf-2 mutants but not in wild-type animals at day 1 of adulthood (Fig. 8A; Fig. S11). At day 8 of adulthood, disruption of drp-1 did not affect mitophagy levels in either wild-type or daf-2 mutants (Fig. 8B; Fig. S11). Mitophagy levels in daf-2;drp-1 worms were also equivalent to daf-2 worms at day 4 of adulthood (Fig. S12). Therefore, disruption of drp-1 further increases mitophagy in daf-2 mutants early in adulthood but has no effect on mitophagy in wild-type animals.

Increased mitophagy resulting from disruption of drp-1 in daf-2 mutants is required for extended longevity. A mtRosella mitophagy reporter strain was used to quantify the effect of drp-1 inhibition on mitophagy in daf-2 worms. At day 1 of adulthood, disruption of drp-1 increases mitophagy in daf-2 mutants but not in wild-type worms (A). This increase in mitophagy is no longer detectable at day 8 of adulthood (B). Inhibiting mitophagy through treatment with RNAi targeting pink-1 significantly decreased the lifespan of daf-2;drp-1 worms but not daf-2 lifespan (C). Combined, these results suggest that an increase in mitophagy contributes to the lifespan extension in daf-2 worms when drp-1 is disrupted. Representative images of mtRosella fluorescence can be found in Fig. S11. Three biological replicates were performed. For lifespan experiments, daf-2 + EV N = 126 daf-2 + pink-1 RNAi N = 84, daf-2;drp-1 + EV N = 91, daf-2;drp-1 + pink-1 RNAi N = 46, temperature = 20 °C. Raw lifespan data can be found in Table S1. Statistical significance was assessed using a two-way ANOVA with Šidák’s multiple comparisons test in panels A and B, and a log-rank test in panel C. Error bars indicate SEM. **p < 0.01,****p < 0.001
To determine whether the early life increase in mitophagy caused by drp-1 disruption contributes to the extended lifespan of daf-2;drp-1 mutants, we treated worms with RNAi targeting pink-1 and measured lifespan, as PINK-1 activation is required for mitophagy to take place. We found that pink-1 RNAi significantly decreased the lifespan of daf-2;drp-1 worms (Fig. 8C). This suggests that the increase in mitophagy caused by disruption of drp-1 is required for drp-1 disruption to increase daf-2 lifespan.
Discussion
In this work, we explored the mechanism by which disruption of drp-1 further extends the lifespan of long-lived daf-2 mutants. We found that disruption of drp-1 acts during development to increase daf-2 lifespan and that drp-1 disruption in neurons, intestine or muscle individually is not sufficient to extend daf-2 longevity. Disruption of drp-1 in daf-2 worms was found to decrease reproduction, enhance resistance to exogenous stressors, increase mitochondrial connectivity, increase peroxisomal connectivity, enhance mitochondrial function and increase mitophagy, all of which may contribute to lifespan extension (Fig. S13). Finally, we found that decreasing mitochondrial fragmentation through targeting other fission genes or RNAi clones that decrease mitochondrial fragmentation independently of drp-1 levels can also extend daf-2 longevity.
Disruption of drp-1 increases mitochondrial and peroxisomal network connectivity
In this work, we show that disruption of drp-1 increases mitochondrial network connectivity in both daf-2 and wild-type young adults. Later in adulthood, disruption of drp-1 continues to generate increased mitochondrial network connectivity, but to a lesser extent. Increased mitochondrial network connectivity as a result of disrupting drp-1 in wild-type animals has previously been documented [35, 40], although we and others have also previously observed no significant difference from wild-type mitochondrial morphology [25, 54, 109].
Using TMRE staining, the mitochondrial network morphology of daf-2 mutants has previously been observed to be altered by disruption of drp-1 at day 3 of adulthood [57]. The ratio of circularity to branching of the mitochondria was quantified and deemed to be higher when drp-1 was disrupted in wild-type, daf-2 and age-1 animals. Here, we similarly observe an increase in mitochondrial circularity, however, we also consistently observe a decrease in the total number of mitochondria, and an increase in the average area of a mitochondrion, indicating that mitochondria are fusing together into larger aggregates. Increased mitochondrial fusion in C. elegans has previously been observed in several long-lived mutants, including daf-2 [34]. Elongated mitochondrial tubules as well as lifespans were reported to be dependent on increased expression of the mitochondrial fusion protein EAT-3.
Given the ability of DRP-1 to regulate peroxisomal fission, we also examined the peroxisomal morphology of daf-2 mutants and found that peroxisomes in the intestine are more interconnected when drp-1 is disrupted. We observed an increase in filamentous structures, as visualized by GFP targeted to peroxisomes and these filaments appeared to connect circular structures together. Filamentous peroxisomal structures have previously been observed in drp-1;fzo-1 worms, which require peroxisomal function for their increased longevity [35]. Thus, morphological changes in both the mitochondrial and peroxisomal network could both contribute to the longevity of daf-2;drp-1 mutants.
Disruption of drp-1 further increases lifespan and stress resistance in daf-2 mutants
In this work, we show that disruption of drp-1 significantly enhances the already increased lifespan and stress resistance of daf-2 mutants. The ability of drp-1 disruption to increase daf-2 and age-1 lifespan was reported previously [57]. We also find that disruption of drp-1 mildly increases wild-type lifespan. This is consistent with findings that disruption of drp-1 can protect against age-associated pathologies in multiple models [55, 110,111,112,113,114], but contrasts with previous findings that drp-1 mutants have a decreased lifespan [54] or have no change in lifespan in C. elegans [35].
To evaluate how disruption of drp-1 affects the healthspan of daf-2 mutants, we compared the thrashing rate of daf-2 and daf-2;drp-1 double mutants at multiple time points. We found that disruption of drp-1 slightly but significantly decreases daf-2 thrashing rate until day 8 of adulthood but does not affect thrashing rate later in adulthood. This data indicates that while disruption of drp-1 may result in a slight decrease of daf-2 motility early in life, it does not markedly affect daf-2 healthspan. Disruption of drp-1 does however significantly affect daf-2 fertility, resulting in a drastic drop in the number of viable progeny produced by each worm. We and others have previously reported that drp-1 mutants also have a significantly lower brood size compared to wild-type [25, 40]. This decrease in fertility could contribute to the ability of drp-1 deletion to extend daf-2 lifespan as many long-lived mutants have decreased fertility and complete ablation of the germline extends longevity [115, 116].
Though it was previously reported that disruption of drp-1 decreases age-1 resistance to chronic oxidative stress by paraquat exposure starting at day 3 of adulthood [57], we find that disruption of drp-1 increases daf-2 resistance to paraquat exposure starting at day 1 of adulthood. Additionally, we find that disruption of drp-1 increases daf-2 resistance to another chronic stress, bacterial pathogen stress by feeding worms Pseudomonas aeruginosa. We have not previously tested the resistance of drp-1 mutants to bacterial pathogen stress, however, we have reported that drp-1 mutants have increased resistance to paraquat exposure and increased activation of the SKN-1-mediated oxidative stress response pathway. We also showed that disruption of other mitochondrial fission genes such as fis-1, fis-2, mff-1 and mff-2 increases resistance to paraquat exposure [25].
Mitochondrial hyper-fusion has been observed in response to oxidative stress in a murine cell model, indicating that increased mitochondrial fusion may be beneficial in response to chronic oxidative stress, perhaps due to mitochondrial complementation mitigating the detrimental effects caused by small amounts of damage. By contrast, we find that disruption of drp-1 decreases daf-2 resistance to oxidative stress by juglone exposure and heat stress, both of which are acute stressors. Disruption of drp-1 also decreases age-1 resistance to heat stress [57]. In comparison, we have previously found that drp-1 mutants have increased resistance to juglone exposure and decreased resistance to heat stress [25]. Thus, the ability of drp-1 disruption to further increase daf-2 resistance to chronic forms of stress may contribute to the ability of drp-1 disruption to extend daf-2 lifespan.
Developmental disruption of drp-1 increases daf-2 lifespan
We find that developmental inhibition of drp-1 is sufficient to increase daf-2 lifespan. This finding is in agreement with our observation that disruption of drp-1 affects daf-2 mitochondrial morphology more significantly in early adulthood compared to later in life and indicates that the mechanism by which inhibition of drp-1 extends daf-2 lifespan takes place during development. Similarly, in long-lived mutants where altered mitochondrial function contributes principally to lifespan extension, genetic inhibition is required during development [88]. By contrast, daf-2 inhibition is required during adulthood, and not during development, for lifespan extension to occur [87]. Thus, while mitochondrial dynamics and the IIS pathway converge to extend lifespan in daf-2;drp-1 mutants, the mechanism by which disruption of drp-1 extends daf-2 lifespan occurs early in life, before disruption of daf-2 contributes to lifespan extension.
Tissue-specific disruption of drp-1 does not increase daf-2 lifespan
Our findings indicate that tissue-specific inhibition of drp-1 in muscles, neurons or intestine fails to increase daf-2 lifespan. This suggests that extension of daf-2 lifespan either requires knockdown of drp-1 in multiple tissues or that tissues not tested here, such as the germline, could be essential for lifespan extension mediated by disruption of drp-1. Inhibition of daf-2 in the intestine or re-expression of daf-16 in daf-2;daf-16 mutants is sufficient to extend longevity [83, 117, 118], while re-expression of daf-2 in the neurons or to a lesser extent the intestines decreases daf-2 lifespan [119]. Combined with our findings here, this suggests that disruption of drp-1 is not acting specifically in the same tissues as the daf-2 mutation to increase lifespan.
In wild-type animals, we found that RNAi inhibition of drp-1 in the neurons extends lifespan. This observation provides a plausible mechanism for why whole-body RNAi inhibition of drp-1 decreases lifespan while deletion of drp-1 increases lifespan. In the deletion mutant, drp-1 is disrupted in all tissues including the neurons and the beneficial effect of drp-1 disruption in neurons mediates the overall increase in lifespan. In wild-type worms treated with RNAi against drp-1, drp-1 will be inefficiently knocked down in the neurons as RNAi is less effective in neuronal tissue [120,121,122]. Without the beneficial effect of disrupting drp-1 in neurons, drp-1 knockdown in the rest of the body has an overall detrimental effect on lifespan. In future studies, it will be interesting to more closely examine the mechanisms by which neuronal knockdown of drp-1 extends lifespan.
Decreasing mitochondrial fragmentation without disrupting drp-1 can extend daf-2 lifespan
We find that inhibiting genes known to be involved in mitochondrial fission as well as genes known to affect mitochondrial fragmentation is sufficient to extend daf-2 lifespan without drp-1 disruption. However, none of the targets tested were able to extend daf-2 lifespan to the same magnitude as drp-1. This could be due to drp-1 disruption having the strongest effect on mitochondrial fragmentation or that another activity of drp-1 also contributes to its effect on daf-2 lifespan.
Of the RNAi clones that decrease mitochondrial fragmentation and increase daf-2 lifespan, we previously reported that inhibition of pgp-3 or alh-12 is able to decrease mitochondrial network disorganization and improve movement in models of polyglutamine toxicity [56]. Furthermore, chemical inhibition of mitochondrial fission has previously been found to increase yeast lifespan during chronological aging [123], another example whereby decreasing mitochondrial fragmentation without inhibiting drp-1 promotes lifespan extension. Limiting drp-1 inhibition by identification of drp-1-alternative mechanisms to promote lifespan extension via decreased mitochondrial fragmentation may be useful given that drp-1 disruption can be detrimental in mammals [124,125,126].
Disruption of drp-1 improves mitochondrial function in young daf-2 adults
Mitochondrial dysfunction and an abnormally high or low mitochondrial membrane potential have both been associated with mitochondrial fragmentation and increased generation of mitochondrial ROS. Under normal physiological conditions, it is thought that increased mitochondrial network fusion promotes a mitochondrial membrane potential that is sufficiently elevated to enhance mitochondrial function without crossing the threshold into ROS overproduction [127]. Decreased membrane potential, or increased ROS levels may act as signals to induce mitochondrial fragmentation and mitophagy [128,129,130].
Though differing outcomes have been reported for the membrane potential of daf-2 mutants [61, 62, 92], increased ATP production and increased ROS generation are consistently observed [62, 63, 90] (Table S5). We find that disruption of drp-1 in daf-2 mutants has no effect on mitochondrial membrane potential or ROS levels but does increase mitochondrial function during early adulthood. Thus, redox homeostasis, which is maintained by DAF-16 and SKN-1 activity in daf-2 mutants [131, 132], remains unaffected by the enhanced mitochondrial function mediated by drp-1 disruption. Increased mitochondrial function in young daf-2;drp-1 mutants, but not later in life, corresponds with our finding that disruption of drp-1 during development is sufficient to extend daf-2 lifespan.
We have previously observed that disruption of drp-1 has no effect on mitochondrial function at day 1 or day 7 of adulthood [25]. Similarly, others have previously reported that drp-1 mutants do not have altered ATP-coupled respiration, although increased basal respiration and an increased proton leak is reported [109]. While drp-1 mutants would be expected to have increased mitochondrial function, membrane potential and mildly elevated ROS levels due to their increased mitochondrial network fusion, in this work, we again show that disruption of drp-1 in wild-type animals does not affect mitochondrial function. Additionally, while we do see a significant increase in ROS levels in drp-1 mutants, we also see a significant decrease in mitochondrial membrane potential. This elevated level of ROS and lowered mitochondrial membrane likely contributes to activation of the SKN-1-mediated oxidative stress response and may explain why disruption of drp-1 is beneficial only under certain conditions. Additionally, the absence of enhanced mitochondrial function may help to explain why disruption of drp-1 in wild-type animals does not extend lifespan as significantly as in daf-2 mutants, where mitochondrial function is enhanced.
Disruption of drp-1 increases induction of mitophagy in young daf-2 adults
Increased DAF-16 and SKN-1 activity leads to increased mitochondrial quality control by selective autophagy in daf-2 mutants [133]. Inhibition of mitophagy partially decreases the lifespan of daf-2 mutants, indicating that increased mitophagy is required to maintain daf-2 longevity [61]. Although DRP-1 is required for selective clearance of dysfunctional mitochondria by recruitment of mitophagy machinery to specific membrane domains, DRP-1-independent mitophagy is possible [20, 134]. However, inhibition of fission machinery has been found to decrease mitophagy, resulting in the accumulation of oxidized mitochondrial proteins and reduced mitochondrial respiration, indicating that mitochondrial fission machinery plays an important role in mitochondrial quality control [21, 135].
Given that inhibiting DRP-1 has previously been found to affect mitophagy activity, we evaluated whether disruption of drp-1 affects mitophagy in daf-2 mutants. We find that disruption of drp-1 further increases daf-2 mitophagy but does not affect wild-type mitophagy levels. Furthermore, as with mitochondrial function, disruption of drp-1 only enhances daf-2 mitophagy in young adults and does not affect mitophagy later in life, corresponding with our finding that disruption of drp-1 during development but not adulthood extends daf-2 lifespan. While increased mitophagy in the absence of DRP-1 would be expected to be excessive and non-selective [20], the enhanced mitochondrial function and maintained ROS and membrane potential seen in daf-2;drp-1 mutants suggest that the increased mitophagy remains beneficial. Importantly, inhibition of mitophagy through pink-1 RNAi prevents drp-1 disruption from increasing daf-2 lifespan suggesting that the increase in mitophagy is contributing to lifespan extension.
Conclusion
Overall, this work defines the conditions under which disruption of drp-1 extends daf-2 lifespan and identifies multiple putative mechanisms that contribute to lifespan extension including enhanced mitophagy. drp-1 inhibition acts during development to extend daf-2 lifespan in multiple tissues or tissues other than intestine, neurons and muscle. Disruption of drp-1 increases mitochondrial and peroxisomal connectedness, decreases reproduction, enhances resistance to chronic stress, increases mitochondrial function and increases mitophagy in daf-2 mutants, all of which may contribute to lifespan extension (Fig. S13). Similar to drp-1, decreasing mitochondrial fragmentation by targeting other mitochondrial fission genes or other genes whose disruption promotes mitochondrial connectivity is sufficient to increase lifespan.
Data availability
Correspondence and material requests should be addressed to Jeremy Van Raamsdonk.
References
Monzel AS, Enriquez JA, Picard M. Multifaceted mitochondria: moving mitochondrial science beyond function and dysfunction. Nat Metab. 2023;5:546–62.
Sharma A, Smith HJ, Yao P, Mair WB. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019;20:e48395.
Lopez-Lluch G. Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech Ageing Dev. 2017;162:108–21.
Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. 2017;74:1999–2017.
Wada J, Nakatsuka A. Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med Okayama. 2016;70:151–8.
Gonzalez-Franquesa A, Patti ME. Insulin resistance and mitochondrial dysfunction. Adv Exp Med Biol. 2017;982:465–520.
Simula L, Nazio F, Campello S. The mitochondrial dynamics in cancer and immune-surveillance. Semin Cancer Biol. 2017;47:29–42.
Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–66.
Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. 2017;8:349–69.
Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res. 2017;1864:1260–73.
Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016;594:509–25.
Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–59.
Srivastava S. The mitochondrial basis of aging and age-related disorders. Genes (Basel). 2017;8:398.
Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):216–31.
Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel). 2017;6:25.
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–18.
Cai Q, Tammineni P. Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci. 2016;10:24.
Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–43.
Scott I, Youle RJ. Mitochondrial fission and fusion. Essays Biochem. 2010;47:85–98.
Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017;216:3231–47.
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46.
Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21–5.
Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–92.
Sato A, Nakada K, Hayashi J. Mitochondrial complementation preventing respiratory dysfunction caused by mutant mtDNA. BioFactors. 2009;35:130–7.
Machiela E, Liontis T, Dues DJ, Rudich PD, Traa A, Wyman L, et al. Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. FASEB J. 2020;34:8475–92.
Wilson PD, Franks LM. The effect of age on mitochondrial ultrastructure. Gerontologia. 1975;21:81–94.
Yasuda K, Ishii T, Suda H, Akatsuka A, Hartman PS, Goto S, et al. Age-related changes of mitochondrial structure and function in Caenorhabditis elegans. Mech Ageing Dev. 2006;127:763–70.
Baumuratov AS, Antony PM, Ostaszewski M, He F, Salamanca L, Antunes L, et al. Enteric neurons from Parkinson’s disease patients display ex vivo aberrations in mitochondrial structure. Sci Rep. 2016;6:33117.
Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–509.
Walter J, Bolognin S, Antony PMA, Nickels SL, Poovathingal SK, Salamanca L, et al. Neural stem cells of Parkinson’s disease patients exhibit aberrant mitochondrial morphology and functionality. Stem Cell Reports. 2019;12:878–89.
Goebel HH, Heipertz R, Scholz W, Iqbal K, Tellez-Nagel I. Juvenile Huntington chorea: clinical, ultrastructural, and biochemical studies. Neurology. 1978;28:23–31.
Liu YJ, McIntyre RL, Janssens GE, Houtkooper RH. Mitochondrial fission and fusion: a dynamic role in aging and potential target for age-related disease. Mech Ageing Dev. 2020;186:111212.
Sgarbi G, Matarrese P, Pinti M, Lanzarini C, Ascione B, Gibellini L, et al. Mitochondria hyperfusion and elevated autophagic activity are key mechanisms for cellular bioenergetic preservation in centenarians. Aging (Albany NY). 2014;6:296–310.
Chaudhari SN, Kipreos ET. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun. 2017;8:182.
Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 2017;26:884.
Zhang Y, Lanjuin A, Chowdhury SR, Mistry M, Silva-Garcia CG, Weir HJ, et al. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. Elife. 2019;8:e49158.
Traa A, Keil A, AlOkda A, Jacob‐Tomas S, Tamez González AA, Zhu S, Rudich Z, Van Raamsdonk JM. Overexpression of mitochondrial fission or mitochondrial fusion genes enhances resilience and extends longevity. Aging Cell. 2024;e14262. https://doi.org/10.1111/acel.14262.
Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J Cell Biol. 2009;186:525–40.
Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, et al. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022.
Breckenridge DG, Kang BH, Kokel D, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008;31:586–97.
Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–26.
Amartuvshin O, Lin CH, Hsu SC, Kao SH, Chen A, Tang WC, et al. Aging shifts mitochondrial dynamics toward fission to promote germline stem cell loss. Aging Cell. 2020;19:e13191.
Ihenacho UK, Meacham KA, Harwig MC, Widlansky ME, Hill RB. Mitochondrial fission protein 1: emerging roles in organellar form and function in health and disease. Front Endocrinol (Lausanne). 2021;12:660095.
Liesa M, Van der Bliek A, Shirihai OS. To fis or not to fuse? This is the question! EMBO J. 2019;38(8):e101839.
Liu R, Chan DC. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol Biol Cell. 2015;26:4466–77.
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–58.
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–62.
Nguyen TT, Voeltz GK. An ER phospholipid hydrolase drives ER-associated mitochondrial constriction for fission and fusion. Elife. 2022;11:e84279.
Basu K, Lajoie D, Aumentado-Armstrong T, Chen J, Koning RI, Bossy B, et al. Molecular mechanism of DRP1 assembly studied in vitro by cryo-electron microscopy. PLoS ONE. 2017;12:e0179397.
Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–56.
Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, Ryan MT. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat Commun. 2018;9:1–15.
Scheckhuber CQ, Erjavec N, Tinazli A, Hamann A, Nystrom T, Osiewacz HD. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol. 2007;9:99–105.
Braun RJ, Westermann B. Mitochondrial dynamics in yeast cell death and aging. Biochem Soc Trans. 2011;39:1520–6.
Traa A, Machiela E, Rudich PD, Soo SK, Senchuk MM, Van Raamsdonk JM. Identification of novel therapeutic targets for polyglutamine diseases that target mitochondrial fragmentation. Int J Mol Sci. 2021;22:13447.
Byrne JJ, Soh MS, Chandhok G, Vijayaraghavan T, Teoh JS, Crawford S, et al. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell Mol Life Sci. 2019;76:1967–85.
Machiela E, Rudich PD, Traa A, Anglas U, Soo SK, Senchuk MM, et al. Targeting mitochondrial network disorganization is protective in C. elegans models of Huntington’s Disease. Aging Dis. 2021;12(7):1753–72.
Yang CC, Chen D, Lee SS, Walter L. The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell. 2011;10:724–8.
van Heemst D, Beekman M, Mooijaart SP, Heijmans BT, Brandt BW, Zwaan BJ, et al. Reduced insulin/IGF-1 signalling and human longevity. Aging Cell. 2005;4:79–85.
Murphy CT, Hu PJ. Insulin/insulin-like growth factor signaling in C. elegans. WormBook: the online review of C. elegans biology. 2013:1–43.
Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277.
Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–8.
Brys K, Castelein N, Matthijssens F, Vanfleteren JR, Braeckman BP. Disruption of insulin signalling preserves bioenergetic competence of mitochondria in ageing Caenorhabditis elegans. BMC Biol. 2010;8:91.
Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556.
Schaar CE, Dues DJ, Spielbauer KK, Machiela E, Cooper JF, Senchuk M, et al. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015;11:e1004972.
Machiela E, Dues DJ, Senchuk MM, Van Raamsdonk JM. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol Dis. 2016;96:1–11.
Palikaras K, Tavernarakis N. In vivo mitophagy monitoring in caenorhabditis elegans to determine mitochondrial homeostasis. Bio Protoc. 2017;7(7):e2215.
Cooper JF, Machiela E, Dues DJ, Spielbauer KK, Senchuk MM, Van Raamsdonk JM. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci Rep. 2017;7:16441.
Koopman M, Michels H, Dancy BM, Kamble R, Mouchiroud L, Auwerx J, et al. A screening-based platform for the assessment of cellular respiration in Caenorhabditis elegans. Nat Protoc. 2016;11:1798–816.
Kirienko NV, Cezairliyan BO, Ausubel FM, Powell JR. Pseudomonas aeruginosa PA14 pathogenesis in Caenorhabditis elegans. Methods Mol Biol. 2014;1149:653–69.
Dues DJ, Andrews EK, Schaar CE, Bergsma AL, Senchuk MM, Van Raamsdonk JM. Aging causes decreased resistance to multiple stresses and a failure to activate specific stress response pathways. Aging (Albany NY). 2016;8:777–95.
Harris-Gauthier N, Traa A, AlOkda A, Moldakozhayev A, Anglas U, Soo SK, et al. Mitochondrial thioredoxin system is required for enhanced stress resistance and extended longevity in long-lived mitochondrial mutants. Redox Biol. 2022;53:102335.
Dues DJ, Schaar CE, Johnson BK, Bowman MJ, Winn ME, Senchuk MM, et al. Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic Biol Med. 2017;108:362–73.
Van Raamsdonk JM, Hekimi S. FUdR causes a twofold increase in the lifespan of the mitochondrial mutant gas-1. Mech Ageing Dev. 2011;132(10):519–21.
Campos JC, Wu Z, Rudich PD, Soo SK, Mistry M, Ferreira JC, et al. Mild mitochondrial impairment enhances innate immunity and longevity through ATFS-1 and p38 signaling. EMBO Rep. 2021;22:e52964.
Wu Z, Isik M, Moroz N, Steinbaugh MJ, Zhang P, Blackwell TK. Dietary restriction extends lifespan through metabolic regulation of innate immunity. Cell Metab. 2019;29(5):1192–205.
Hahm JH, Kim S, DiLoreto R, Shi C, Lee SJ, Murphy CT, et al. C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nat Commun. 2015;6:8919.
Roy C, Molin L, Alcolei A, Solyga M, Bonneau B, Vachon C, et al. DAF-2/insulin IGF-1 receptor regulates motility during aging by integrating opposite signaling from muscle and neuronal tissues. Aging Cell. 2022;21:e13660.
Evans EA, Chen WC, Tan MW. The DAF-2 insulin-like signaling pathway independently regulates aging and immunity in C. elegans. Aging Cell. 2008;7:879–93.
Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13(11)1385–93.
McColl G, Rogers AN, Alavez S, Hubbard AE, Melov S, Link CD, et al. Insulin-like signaling determines survival during stress via posttranscriptional mechanisms in C. elegans. Cell Metab. 2010;12:260–72.
Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, et al. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics. 1998;150:129–55.
Tissenbaum HA, Ruvkun G. An insulin-like signaling pathway affects both longevity and reproduction in Caenorhabditis elegans. Genetics. 1998;148:703–17.
Zhang YP, Zhang WH, Zhang P, Li Q, Sun Y, Wang JW, et al. Intestine-specific removal of DAF-2 nearly doubles lifespan in Caenorhabditis elegans with little fitness cost. Nat Commun. 2022;13:6339.
Dues DJ, Andrews EK, Senchuk MM, Van Raamsdonk JM. Resistance to stress can be experimentally dissociated from longevity. J Gerontol A Biol Sci Med Sci. 2019;74:1206–14.
Soo SK, Traa A, Rudich ZD, Moldakozhayev A, Mistry M, Van Raamsdonk JM. Genetic basis of enhanced stress resistance in long-lived mutants highlights key role of innate immunity in determining longevity. Aging Cell. 2023;22:e13740.
Wu Z, Senchuk MM, Dues DJ, Johnson BK, Cooper JF, Lew L, et al. Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018;16:147.
Dillin A, Crawford DK, Kenyon C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science. 2002;298:830–4.
Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298(5602):2398–401.
Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem. 2008;143:449.
Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–65.
Wei Y, Kenyon C. Roles for ROS and hydrogen sulfide in the longevity response to germline loss in Caenorhabditis elegans. Proc Natl AcadSci U S A. 2016;113:E2832-2841.
Lemire BD, Behrendt M, DeCorby A, Gaskova D. C. elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech Ageing Dev. 2009;130:461–5.
Rottenberg H. The reduction in the mitochondrial membrane potential in aging: the role of the mitochondrial permeability transition pore. Int J Mol Sci. 2023;24:12295.
Berry BJ, Vodickova A, Muller-Eigner A, Meng C, Ludwig C, Kaeberlein M, et al. Optogenetic rejuvenation of mitochondrial membrane potential extends C. elegans lifespan. Nat Aging. 2023;3:157–61.
Berry BJ, Mjelde E, Carreno F, Gilham K, Hanson EJ, Na E, et al. Preservation of mitochondrial membrane potential is necessary for lifespan extension from dietary restriction. Geroscience. 2023;45:1573–81.
Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091-13096.
Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med. 2011;32:159–221.
Iser WB, Wolkow CA. DAF-2/insulin-like signaling in C. elegans modifies effects of dietary restriction and nutrient stress on aging, stress and growth. PLoS One. 2007;2:e1240.
Shoyama T, Shimizu Y, Suda H. Decline in oxygen consumption correlates with lifespan in long-lived and short-lived mutants of Caenorhabditis elegans. Exp Gerontol. 2009;44:784–91.
Macedo F, Romanatto T, Gomes de Assis C, Buis A, Kowaltowski AJ, Aguilaniu H, et al. Lifespan-extending interventions enhance lipid-supported mitochondrial respiration in Caenorhabditis elegans. FASEB J. 2020;34:9972–81.
Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–6.
Maglioni S, Mello DF, Schiavi A, Meyer JN, Ventura N. Mitochondrial bioenergetic changes during development as an indicator of C. elegans health-span. Aging (Albany NY). 2019;11:6535–54.
Van Raamsdonk JM, Meng Y, Camp D, Yang W, Jia X, Benard C, et al. Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics. 2010;185:559–71.
Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361.
Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–44.
Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010;9:433–47.
Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016; 22(8):879–88.
Srivastava V, Gross E. Mitophagy-promoting agents and their ability to promote healthy-aging. Biochem Soc Trans. 2023;51:1811–46.
Luz AL, Rooney JP, Kubik LL, Gonzalez CP, Song DH, Meyer JN. Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PloS One. 2015;10(6):e0130940.
Duan C, Wang L, Zhang J, Xiang X, Wu Y, Zhang Z, et al. Mdivi-1 attenuates oxidative stress and exerts vascular protection in ischemic/hypoxic injury by a mechanism independent of Drp1 GTPase activity. Redox Biol. 2020;37:101706.
Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, et al. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun. 2014;5:5244.
Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, et al. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1458.
Yan J, Liu XH, Han MZ, Wang YM, Sun XL, Yu N, et al. Blockage of GSK3β-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol Aging. 2015;36:211.
Manczak M, Kandimalla R, Fry D, Sesaki H, Reddy PH. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Human Mol Genet. 2016;25(23):5148–66.
Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 1999;399:362–6.
Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C. Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science. 2002;295:502–5.
Uno M, Tani Y, Nono M, Okabe E, Kishimoto S, Takahashi C, et al. Neuronal DAF-16-to-intestinal DAF-16 communication underlies organismal lifespan extension in C. elegans. iScience. 2021;24:102706.
Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502.
Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science. 2000;290:147–50.
Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:RESEARCH0002.
Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–12.
Asikainen S, Vartiainen S, Lakso M, Nass R, Wong G. Selective sensitivity of Caenorhabditis elegans neurons to RNA interference. NeuroReport. 2005;16:1995–9.
Palermo V, Falcone C, Calvani M, Mazzoni C. Acetyl-L-carnitine protects yeast cells from apoptosis and aging and inhibits mitochondrial fission. Aging Cell. 2010;9:570–9.
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–66.
Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–16.
Yamamori T, Ike S, Bo T, Sasagawa T, Sakai Y, Suzuki M, et al. Inhibition of the mitochondrial fission protein dynamin-related protein 1 (Drp1) impairs mitochondrial fission and mitotic catastrophe after x-irradiation. Mol Biol Cell. 2015;26:4607–17.
Jezek J, Cooper KF, Strich R. Reactive oxygen species and mitochondrial dynamics: the Yin and Yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel). 2018;7(1):13.
Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–9.
Galloway CA, Yoon Y. Perspectives on: SGP symposium on mitochondrial physiology and medicine: what comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol. 2012;139:455–63.
Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–51.
Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–38.
Tullet JMA, Green JW, Au C, Benedetto A, Thompson MA, Clark E, et al. The SKN-1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell. 2017;16:1191–4.
Princz A, Pelisch F, Tavernarakis N. SUMO promotes longevity and maintains mitochondrial homeostasis during ageing in Caenorhabditis elegans. Sci Rep. 2020;10:15513.
Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol. 2016;215:649–65.
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80.
Acknowledgements
Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P30 OD010440). We would also like to acknowledge the C. elegans knockout consortium and the National Bioresource Project of Japan for providing strains used in this research.
Funding
This work was supported by the Canadian Institutes of Health Research (CIHR; http://www.cihr-irsc.gc.ca/; JVR; Application 399148 and 416150) and the Natural Sciences and Engineering Research Council of Canada (NSERC; https://www.nserc-crsng.gc.ca/index_eng.asp; JVR; Application RGPIN-2019–04302). JVR is the recipient of a Senior Research Scholar career award from the Fonds de Recherche du Québec Santé (FRQS) and Parkinson Quebec. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: AT, JVR. Methodology: AT, ATG, JVR. Investigation: AT, ATG. Analysis: AT, ATG, JVR. Visualization AT, ATG, JVR. Writing – original draft: AT. Writing – review and editing: AT, ATG, JVR. Supervision: JVR.
Corresponding author
Ethics declarations
Disclosures
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Traa, A., Tamez González, A.A. & Van Raamsdonk, J.M. Developmental disruption of the mitochondrial fission gene drp-1 extends the longevity of daf-2 insulin/IGF-1 receptor mutant. GeroScience (2024). https://doi.org/10.1007/s11357-024-01276-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11357-024-01276-z