
Abstract
The presence of PFAS in drinking water may pose a serious threat to human health. This study aims to determine the levels of these compounds and their precursors in water samples from a drinking water treatment plant (DWTP) located in l’Ampolla (Spain) and to assess their fate. Additionally, ten Spanish bottled waters were analyzed to compare the occurrence of PFAS in the mentioned matrices and in drinking water. Off-line solid phase extraction (SPE) followed by liquid chromatography-tandem mass spectrometry (HPLC–MS/MS) was applied to determine 26 PFAS and PFAS precursors after a total oxidizable precursor assay. The analytical method presents low quantification limits (0.25–5 ng/L). A total PFAS concentration of up to 16 ng/L in all the DWTP samples was obtained, and 6:2 FTS was the only precursor detected. Results are close to the quantification limits, resulting in a high degree of uncertainty, and for this, it is difficult to evaluate the DWTP PFAS removal efficiency. Regarding bottled water, total PFAS concentration found was up to 12 ng/L in one of ten samples, with no precursors detected. Exposure assessment revealed that there is no risk associated with the ingestion of the samples analyzed. Moreover, there were no differences in terms of risk between drinking water from l’Ampolla DWTP and bottled water.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Per- and polyfluoroalkyl substances (PFAS), known as “forever chemicals” are a wide group of more than 7 million chemicals, including salts and mixtures. These compounds consist of a hydrophobic alkyl chain of variable length depending on the number of carbons (European Environment Agency 2022; Schymanski et al. 2023). This carbon chain could be totally or partially fluorinated, ending with a hydrophilic group. Their properties make PFAS useful in a wide range of processes and applications such as paints, textiles, and food packaging (Glüge et al. 2020; The Interestate Technology and Regulatory Council 2023). Due to their large-scale production and their widespread use, PFAS have reached the environment and tend to bioaccumulate in wildlife and human beings (Lasier et al. 2011; Lee et al. 2020; Poothong et al. 2020). Nowadays, several studies reveal negative effects on reproductive toxicity, neurotoxicity, immunotoxicity, and carcinogenicity, among others (Pizzurro et al. 2019; Pilli et al. 2021), which provide fundamentals for regulatory authorities to regulate PFAS compounds. Driven by different reports on toxic effects, in 2008, the European Food Safety Authority (EFSA) established, and recently revised, a tolerable weekly intake (TWI) for the sum of perfluorooctanoic acid (PFOA), perfluorooctanesulfonic acid (PFOS), perfluorononanoic acid (PFNA), and perfluorohexanesulfonic acid (PFHxS) with a value of 4.4 ng/kgbw per week (Schrenk et al. 2020).
Drinking water is one of the routes of human exposure to PFAS through dietary intake, other being dust, indoor air inhalation, and dermal contact (Poothong et al. 2020). Because of that, PFAS is a topic of concern in the scientific community, and several research studies related to occurrence, fate in environment, and in drinking and wastewater treatment plants, as well as on human health exposure to these compounds in surface and drinking water, have been published to date. The occurrence of PFAS in surface water could be an indicator of their potential presence in drinking water (McMahon et al. 2022) since some studies in the literature suggested that, in general, drinking water treatment plants (DWTP) applying conventional treatment steps like coagulation, flocculation, sedimentation, and filtration do not tend to effectively remove PFAS, even though other studies have demonstrated that advanced treatment processes like activated carbon, ion exchange resins, nanofiltration, and reverse osmosis may be effective in removing these compounds (Rahman et al. 2014; Sun et al. 2016; Kim et al. 2020).
Although PFAS generally occur at low concentrations in environmental waters, their occurrence and risk is a growing worldwide concern, and in this sense, these compounds have been regulated in the European Drinking Water Directive, with a limit of 500 ng/L for all PFAS (Organization for Economic Cooperation and Development (OECD) 2023). This Directive has been transposed into Spanish legislation, in particular in Royal Decree 3/2023, where the parametrical value for all PFAS is 100 ng/L; a parametrical value of 70 ng/L for perfluorobutanoic acid (PFBA), PFHxS, PFOA and PFNA, individually, has been also established (Ministerio de la Presidencia R con las C y MD 2023).
The increasing concern regarding PFAS substances in water samples implies the need for the development of analytical methods and, consequently, different approaches to determine PFAS in water have been reported in recent years by several authors (Schultz et al. 2006; Ciofi et al. 2018; Borrull et al. 2020; Mottaleb et al. 2021). All of these are generally based on liquid chromatography (HPLC) coupled with triple quadrupole (QqQ), time-of-flight (QTOF), and Quadrupole-Orbitrap mass spectrometry, as Teymoorian et al (2022) presented in their recent research review work.
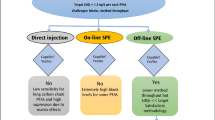
There are different strategies currently in use for sample pretreatment. While some authors use direct injection or large volume injection of water samples (between 0.1 and 0.9 mL Ciofi et al. 2018; Borrull et al. 2020; Mottaleb et al. 2021)), the most common strategy is to include an SPE procedure (between 100 and 5000 mL (Teymoorian et al. 2022)). Direct injection of water samples is used in some cases but, even though this is a quick way to determine PFAS (Schultz et al. 2006; Ciofi et al. 2018; Borrull et al. 2020; Mottaleb et al. 2021), solid phase extraction (SPE) is the most used strategy to be able to reach the usual PFAS levels in environmental water samples (sub-ng/L levels). Regarding SPE cartridge types, the most used is anion exchange mix-mode sorbent > weak anion exchange sorbent > lipophilic-hydrophilic balance sorbent (HLB) (Teymoorian et al. 2022), and the reported recoveries are in the range of 70–110% for most of the carboxylic and sulfonate PFAS (Kaboré et al. 2018; Coggan et al. 2019; Chen et al. 2021; Jurikova et al. 2022).
The scientific community is also working on determining PFAS precursors, also referred to as “hidden PFAS.” This is a challenging task for various reasons, as the precursors are unknown, they may degrade, and there are no available standards (Al Amin et al. 2020). Given this, total oxidizable precursor (TOP) assay is applied—with persulfate and basic conditions—and it is based on the oxidation of PFAS precursors into perfluoroalkyl acids (PFAAS), which can be easily determined by the HPLC–MS/MS target method. Knowing the PFAS concentration before and after TOP, it is then possible to assess the total concentration of PFAS precursors in samples (Houtz and Sedlak 2012; Martin et al. 2019; Nolan et al. 2019; Kaiser et al. 2021; Nikiforov 2021). This assay has been applied to different environmental matrices such as soil, biota, or water. For example, Martin et al. (2019) reveals the presence of PFAS precursors such as 6:2 fluorotelomer sulfonamide alkylbetaine (6:2 FTAB), 6:2 fluorotelomer sulfonate (6:2FTS), 8:2 fluorotelomer sulfonate (8:2FTS), perfluorooctanesulfonamide (PFOSAm), and perfluorohexanesulfonamide (FHxSA) in groundwater samples from fire-equipment testing sites in Canada after performing TOP.
The above-mentioned analytical methodologies have been widely applied to monitor PFAS presence in drinking waters. In this sense, elevated concentrations of PFAS (between 20 and 20,000 ng/L) have been reported, particularly near industrial or fire-training activities sites (Mussabek et al. 2023). On the contrary, PFAS in drinking water worldwide ranges from 0.08 to 11.27 ng/L (Turkey) and to 300 ng/L (Czech Republic) in terms of total concentration and from 0.48 ng/L (Turkey) to 18.9 ng/L (USA) in bottled water (Teymoorian et al. 2022).
Given the context presented above, the aim of the present study is to apply a method based on off-line SPE HPLC–MS/MS to determine 26 PFAS at ng/L levels in selected DWTP water samples from different treatment processes as well as surface water (Ebro River water) and drinking water. DWTP water samples were collected to assess PFAS fate and to analyze the removal effectiveness of a DWTP equipped with advanced treatment processes. Moreover, another aim was to have an overview of the PFAS contamination status in bottled water, as it is an important source of drinking water in Spain, and to compare the occurrence of these compounds in the mentioned matrix and in drinking water. Total oxidizable assay (TOP) was performed to have an estimation of the number of possible PFAS precursors (non-target compounds in the analytical method) present in samples as they can contribute to increasing the total final PFAS concentration. Finally, an assessment has been performed of exposure and human health risk from targeted PFAS due to ingestion of l’Ampolla DWTP drinking water and bottled water.
Materials and methods
Reagents and standards
High purity chemical standards of target PFAS were purchased from AccuStandard (New Haven, USA) (a mix standard of 24 PFAS) and Wellington Laboratories (Whitby, ON, Canada) (two individual PFAS). Moreover, a mix standard of 13 isotopically labelled PFAS was obtained from Wellington Labs, Inc. (Whitby, ON, Canada) and was used as surrogate standards. The 26 target and 13 isotopically labelled PFAS acronyms, retention time, and MRM mass transitions are shown in Table 1.
A working standard solution containing 26 target PFAS was prepared with methanol (MeOH) to a final concentration of 5000 ng/L and then stored at − 20 °C in amber glass vials. The same procedure was applied to get a working standard solution of isotopically labelled PFAS to a final concentration of 5000 ng/L.
HPLC water, MeOH, acetonitrile (ACN), ammonium acetate, and formic acid were of HPLC grade and were obtained from Honeywell (Seelze, Germany). Potassium persulfate (> 99%) was acquired from Sigma-Aldrich (St. Louis, MO, USA). Sodium hydroxide (NaOH) was acquired from J.T. Baker (Gliwice, Poland), and hydrochloric acid (HCl) was obtained from Supelco (Darmstadt, Germany).
Bond Elut PFAS WAX SPE cartridges (150 mg/6 mL, 50 µm) purchased from Agilent Technologies (USA) were used for the extraction procedure, as well as regenerated cellulose syringe filters (13 mm Ø, 0.45 µm) from LLG Labware (USA).
Sample collection and preparation
Water samples were collected, each month, from l’Ampolla DWTP between January and November 2022. This DWTP, with the capacity to treat up to 4 m3/s of water, comprises the following treatment steps: CO2 pH adjustment, pre-ozonation, coagulation-flocculation-decantation, sand filtration, post-ozonation, granular-activated carbon filtration (GAC), ultra-violet disinfection (UV), and NaClO chlorination. Figure 1, which is a schematic of l’Ampolla DWTP processes, shows the sampling points established in the present study: (1) at DWTP influent (water from Ebro River), (2) after pre-ozonation, (3) after post-ozonation, and (4) at DWTP drinking water (after final chlorination with NaClO).

Schematic of the DWTP processes. 1 to 4 are the selected sampling points
Water samples were collected in 500 mL high density polyethylene (HDPE) plastic bottles. After collection, all samples were stored at 3 ± 2 °C until analysis.
Ten bottled water samples from different commercial brands were purchased from local supermarkets of l’Ampolla, Tarragona. After that, samples were also stored at 3 ± 2 °C until analysis.
An intercomparison sample with 32 native compounds was obtained from Round 306 of ERA’s Water Supply for Drinking Water PT Scheme.
SPE method
Before assay, each sample was divided in two. One half was used for determining 26 target PFAS by SPE method and the other half to perform TOP assay. All samples were analyzed in triplicate.
The extraction method was based on off-line SPE. One hundred milliliters of sample, after the addition of the mixture of isotopically labelled surrogates in methanol and its acidification with 1.25 mL of formic acid (pH < 3), was loaded onto the Agilent Bond Elut PFAS WAX cartridge. The cartridges were pre-conditioned with 5 mL of 3% NH4OH in 60/40 ACN/MeOH, 5 mL of HPLC water with 0.3 M of formic acid. Then, samples were loaded into cartridges and rinsed with 5 mL HPLC water. Finally, target analytes were eluted into Falcon tubes with 5 mL of 3% NH4OH in 60/40 ACN/MeOH. The extract was evaporated using a vacuum centrifugal concentrator, IR Micro-Cenvac (N-Biontek, Korea) to a final volume of 500 µL (Hunt et al. 2018). Finally, it was adjusted to 1 mL with ACN:H2O (1:1) and filtered using a 0.22-µm cellulose regenerated syringe filter before injection.
TOP-SPE method
TOP assay was conducted according to Houtz and Sedlak (2012) and Martin et al. (2019), with some modifications. Briefly, a 100 mL water sample aliquot was placed in a 100 mL HDPE bottle and a final concentration 0.18 M of potassium persulfate and 0.45 M of NaOH was added. Samples were heated in a water bath for 6 h at 85 °C, shaking it regularly. The pH of each sample was measured before and after sample heating to confirm it was maintained during TOP assay (pH > 12). After that, samples were cooled down to ambient temperature in an ice bath, and pH was brought to approximately 8 with HCl to stop the reaction. Then, the final extract was acidified with formic acid (pH < 3) and subjected to SPE as has been explained in the “SPE method” section.
Equipment and chromatographic conditions
The analysis of the obtained extracts was performed using an Agilent chromatographic system (Agilent Technologies, Waldbronn, Germany) equipped with a 1260 HiP degassing unit, 1260 binary pump, 1260 multisampler, and a 1260 thermostated column compartment. The target PFAS were chromatographically separated with a Poroshell 120 EC-C18 analytical column (50 mm × 3 mm, 2.7 µm) and a 120 EC-C18 guard cartridge (5 mm × 3 mm × 2.7 µm). Water with 10 mM ammonium acetate (A) and ACN (B) was used as mobile phase. The gradient profile started at 10% B, which was raised to 40% B within 2 min, then to 95% B in 4 min and then 100% B in 6.5 min. Finally, it returned to initial conditions (10% B) in 0.5 min and held for 5 min to equilibrate the column for the next analysis. A flow rate of 0.40 mL/min was used and the temperature of the chromatographic column was kept at 45 °C. The injection volume was 100 µL. The acquisition was performed in dynamic multiple reaction monitoring (dMRM) in negative mode using an ESI source. The optimal conditions for the analytes were as follows: a nitrogen flow rate of 8 mL/min, a nebulizer pressure of 30 psi (N2) and source temperature of 300 °C. The collision energies for all compounds were from 4 to 200 V. The optimal values of HPLC–MS/MS parameters are summarized in Table 1.
To reduce background contamination, an analytical LC column (Poroshell 120 EC-C18, 100 mm × 4.6 mm × 2.7 µm) was installed between the mobile phase mixing chamber and the sample injector as a delay column. Moreover, all polyeterketone tubes (PEEK) were substituted by stainless steel ones.
Method performance evaluation
The validation of this method followed the EPA validation guideline for PFAS method (United States Environmental Protection Agency 2018), covering several aspects including linearity, detection and quantification limits (LOD and LOQ), repeatability, accuracy, matrix effect, recovery, and trueness which was evaluated through z-scores. Analogue isotopically labelled standards (ISTD) were used for almost all target compounds. However, for some compounds where the analogue ISTD was unavailable, surrogate-corrected recovery of the extraction procedure and surrogate-corrected matrix effect were assessed.
The SPE extraction step recovery (% RE) was calculated as the ratio of the peak areas for a sample spiked at a concentration of 100 ng/L of target analytes and 50 ng/L of ISTD before SPE extraction compared to the ratio of the peak areas obtained from the sample spiked at the same concentrations after SPE.
For some PFAS compounds, no isotopically labelled standards analogues were available. For this reason, their quantification has been performed by the interpolation of the signal in the calibration curve (without using ISTD).
The surrogate-corrected matrix effect (% ME) was also assessed. It was calculated as the ratio between the analyte signal response obtained from the spiked sample matrix compared with that obtained when a neat sample is spiked. Matrix effect factor is considered acceptable when it is between the range of 20 and − 20%.
Limits of detection (LODs) correspond to the concentration whose signal-to-noise (S/N) ratio is greater than 3, and limits of quantification (LOQs) were defined as the lowest point of the calibration curve with S/N ≥ 10.
The repeatability of the method was expressed as relative standard deviation (% RSD) of five replicates at a concentration of 5 ng/L, 50 ng/L, and 100 ng/L.
TOP assay has been performed to assess the presence of PFAS precursors in water samples. It is necessary to evaluate the oxidation efficiency of the applied assay by spiking surface water from the Ebro River with 6:2 FTS (which is a PFAS precursor). Briefly, 10 µL of 1 µg/mL of 6:2FTS standard was added to a 100 mL polyethylene bottle and evaporated to dryness without heating. After that, it was redissolved with 100 mL of surface water, and it was subjected to the TOP-SPE method (“TOP-SPE method” section).
Exposure and human health risk assessment
The presence of PFAS in drinking water turns water ingestion into a potential exposure route for these compounds to reach our organism. Thus, in 2020, the EFSAs CONTAM Panel (Panel on Contaminants in the Food Chain), decided to consider the sum of PFOA, PFNA, PFHxS, and PFOS as main contributors when estimating the possibility of these compounds causing adverse health effects by calculating the estimated weekly intake (EWI).
where ∑CPFOA, PFNA, PFHxS, PFOS represents the sum of the average measured concentrations in ng/L (considering LOQs for non-detected compounds), DWI (L/day) is the drinking water intake (2 L/day for adults, 1 L/day for children and 0.75 L/day for bottled-fed infants), FOC is the frequency of water consumption (7 day/week), and BW (kg) represents body weight (60 kg for adults, 10 kg for children, and 5 kg for bottled-fed infants). The DWI and BW data were obtained from the WHO Guidelines for drinking water quality (World Health Organization 2022).
For the health risk assessment, hazard quotients (HQ) for the aforementioned four PFAS were calculated according to Eq. (2) (Dvorakova et al. 2023):
where RfD is the reference dose, considered as TWI for ∑4 PFAS = 4.4 ng/kgbw/week and is related to the oral exposure to the human population without appreciable risk of negative effects during lifetime exposure.
The interpretation of the HQ value obtained is as follows: HQ < 0.1, indicates no hazard; HQ = 0.1–1.0, indicates a low hazard; HQ > 1–10, indicates a moderate hazard; HQ > 10 indicates a high hazard.
Results and discussion
Method validation
The presented analytical method was successfully validated with the parameters described in the “Method performance evaluation” section by SPE and HPLC–MS/MS using drinking water and Ebro River water. The extraction procedure, surrogate-corrected recovery (% RE), surrogate-corrected matrix effect (% ME), as well as linearity, LODs, and LOQs, and the repeatability were evaluated.
The surrogate-corrected % RE and % ME were evaluated for 100 mL of drinking water and Ebro River water using samples spiked with targeted PFAS at 100 ng/L and ISTD at 50 ng/L. However, for some target compounds, there was no availability of the analogue ISTD. The results for these two parameters are shown in Table 2. The % RE of almost all analytes in both matrices are similar and fall within the 70 to 130% range required by EPA methods (United States Environmental Protection Agency 2018). In the case of N-EtFOSAA, PFUnS, and PFTrS, surrogate-corrected recoveries in both matrices were not considered acceptable despite the use of non-analogue ISTD. In these specific situations, their quantification has been performed by the interpolation of the signal in the calibration curve. The results obtained were satisfactory and useful for studying possible analyte losses or contamination during sample preparation. Surrogate-corrected matrix effect was calculated as the ratio of the signal of each analyte spiked in the Ebro River water and drinking water after the SPE extraction and their signal for standard sample solutions. As shown in Table 2, most compounds have |% ME|< 20%, indicating that the matrix effect is not significant and that good recoveries were achieved from both water sample matrices. However, for 4:2 FTS, 8:2 FTS, N-MetFOSAA, and N-EtFOSAA surrogate-corrected |% ME| exceeded 20%, possibly due to the use of non-analogue ISTD. In these situations, % ME are not considered acceptable, and as explained before, their quantification has been performed by the interpolation of the signal in the calibration curve.
Curves were plotted, and the linear ranges of calibration for each target PFAS compound are shown in Table 3. Linearity was good for all the compounds (R2 > 0.996), except for 6:2 FTS, N-MetFOSAA, N-EtFOSAA, PFDS, and PFTrS, which coefficients ranged between 0.93 and 0.98 due to the dispersion of the obtained results. The LODs were between 0.1 and 1.7 ng/L and LOQs (the lowest point of each calibration curve) were between 0.25 and 5 ng/L for both matrices because the % RE and % ME were similar in both cases. Taking into account current guidelines (Ministerio de la Presidencia R con las C y MD 2023), the obtained limits were satisfactory to assess the presence of the mentioned analytes in drinking water. The uncertainty of the measurement was 40%, which is lower than the requirement specified in the Spanish directive (RD 3/2023).
The method accuracy met the expected criteria (70–130%) at three concentration levels (5, 50, and 100 ng/L) and as well as precision with RSD < 20% (n = 5). These results were comparable to those obtained with other recent methods (Coggan et al. 2019; Martin et al. 2019; Liu et al. 2020).
In the ERA proficiency test, 18 target analytes were determined in drinking water at concentrations ranging from < 60 to 486 ng/L (the intercomparison sample was diluted because the concentration exceeded 100 ng/L). The trueness of the obtained results was assessed using the z-score parameter for each analyte, achieving values from − 1.13 to 1.26, within the satisfactory interval of − 2 to 2, indicating the reliability of the presented method.
Overall, based on the validation parameters of the method, we can conclude that this method is suitable for the quantification of the PFAS under study in drinking and surface water within in the working ranges of 0.25–5 ng/L and 5–100 ng/L.
Analysis of targeted PFAS
Occurrence in DWTP water samples
Twenty-six PFAS compounds were determined in water samples from different treatment processes of l’Ampolla DWTP per triplicate. Sampling locations were selected taking into account information from different studies in the bibliography that demonstrated that oxidation processes involving the use of ozone, GAC, and UV could decrease, enhance, or be ineffective in terms of PFAS concentration (Takagi et al. 2011; Rahman et al. 2014; Thomas et al. 2020; Verma et al. 2021a; Zhong et al. 2023).
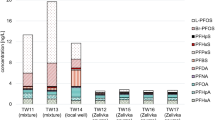
Of 26 PFAS compounds monitored in the present study (20 of them included in RD 3/2023 to be regulated until January 2025), 17 were detected at least once in water samples from different water treatment stages at the l’Ampolla DWTP (Ebro River water, preozonation, postozonation, and drinking water), with detection frequencies of 25–100% (Table 4). As shown in Fig. 2, the ∑20 PFAS average concentration in samples from the DWTP ranged between 11 and 16 ng/L, which results are lower than 100 ng/L, the parametrical value specified in Spanish legislation (Ministerio de la Presidencia R con las C y MD 2023). Moreover, PFOA, PFNA, PFHxS, and PFOS concentrations in drinking water were 0.3 ng/L, 1.1 ng/L, and < LOQ for PFHxS and PFOS, respectively, all below the individual parametrical value of 70 ng/L for each compound) (Ministerio de la Presidencia R con las C y MD 2023). Regarding DWTP influent water, the obtained results (1.7 ng/L for PFBA, 0.8 ng/L for PFBS, 0.4 ng/L for PFOA, and < LOQ for PFOS) are in line with a study of the literature regarding PFAS content in samples from areas without industrial influence. In this case, the results obtained were < 5 ng/L for PFOA and < LOQ (0.6 ng/L) for PFBS, PFDA, PFNA, PFOS, and PFTeA (Castiglioni et al. 2015).

Total (∑20) PFAS average concentration in Ebro River water, pre-ozonation sample, post-ozonation sample, drinking water, and bottled water
The behavior of PFAS in the DWTP processes was investigated. In general, PFAS concentration (taken as the average for the studied period) decreased by approximately 30% between the DWTP effluent water and influent water of the same plant. This reduction can be attributed to the different treatment processes that take place there. In this study, the DWTP ozone dosage varied between 0.4 and 0.6 g/m3, with water pH in pre- and post-ozonation stages maintained at 7.5. These conditions could potentially oxidize the present PFAS compounds, as similar operation conditions in the literature have shown an increase in PFAS concentration after pre-ozonation (0.5 g/m3) (Verma et al. 2021b). Nonetheless, as observed in Fig. 2, water samples collected after the pre- and post-ozonation steps show minimal variations in PFAS concentrations under typical DWTP conditions. However, the values for individual PFAS activities obtained are close the LOQ, which must be considered when assessing PFAS removal/oxidation at this stage. After post-ozonation, the treated water entered the GAC unit. In general, long-chain PFAS are removed in the GAC unit due to their high hydrophobicity, while shorter PFAS are less effectively removed (Rahman et al. 2014; Wang et al. 2020). In L’Ampolla DWTP, water enters a UV-disinfection process (λ = 254 nm) after the GAC unit. As observed in Table 4, both the influent and effluent of the DWTP have low or no concentrations of long-chain PFAS. However, short-chain PFAS were detected in the influent but were not removed by GAC. The PFAS compound concentrations found in the surface water used as DWTP raw water were at low ng/L levels and, at the specific time when the study was conducted, the activated carbon was aged (Rahman et al. 2014). For these reasons, it is not possible to appreciate the removal efficiency of the GAC followed by the UV-disinfection process (λ = 254 nm) conclusively in this study.
Occurrence in bottled water samples
As regards bottled waters, ten natural mineral bottled waters, contained in polyethylene terephthalate bottles, from different Spanish sources were analyzed. Only 7 PFAS compounds were positively detected, with concentrations below LOQ (PFBA, PFBS, PFHxA, PFHpA, PFHpS, and PFDoA) in 9 of the ten samples analyzed. However, as shown in Fig. 2, one sample showed the presence of 0.5 ng/L for PFOA, 0.4 ng/L for PFNA, 2.7 ng/L for PFNS, 0.5 ng/L for PFDA, 3.6 ng/L for PFDS, and 0.5 ng/L for PFDoA (RSD% < 15% for n = 3) total PFAS content being 8 ng/L. The quantified PFAS compounds have analogue ISTD, so no correction has been applied in terms of matrix effect. The concentration of PFAS in bottled water varies between studies from different countries around the world, from < LOQ to 15 ng/L, which are results in line with those presented in this study, and in general lower than those obtained for drinking water (Teymoorian et al. 2022). The differences in PFAS concentrations between drinking water from a DWTP and bottled water are probably due to water origin. In this case, drinking water is produced by treating river water, which can be influenced by industrial or wastewater treatment plant effluents, while bottled water typically comes from groundwater sources where PFAS contamination is limited.
Analysis of PFAS precursors
One consideration that should be taken into account when performing a TOP assay is the evaluation of the oxidation efficiency, as explained in the “Method performance evaluation” section. In a first attempt, using the same doses of oxidant as used by Houtz and Sedlak (2012), the presence of 6:2 FTS was observed after TOP assay, which indicated an incomplete oxidation, and which implies an underestimation of precursor concentration. This fact may be related to the presence of co-contaminants, sample organic content, halide ions, carbonates, or humic acid (Ateia et al. 2023). As shown in Table 5, two different oxidant dosages conditions were tested. In both cases, no precursor was determined after the TOP assay, and the ratio between the sum of concentrations of PFAA precursors and the sum of total PFAS was less than 5%, which indicated a successful oxidation (Nolan et al. 2019). Moreover, the molar conversion yields of 6:2 FTS obtained were in line with those from the literature. One hundred eighty millimolar of persulfate and 450 mM of NaOH were the suitable dosage that fully oxidized 6:2 FTS in the surface water under study.
After that, the presence of PFAS precursors was evaluated. In the presented analytical method, some precursors have been included as targeted compounds (4:2 FTS, 6:2 FTS, 8:2 FTS, PFOSA, N-MetFOSAA, and N-EtFOSAA) and only one of them, 6:2 FTS, was detected in L’Ampolla water samples at < LOQ concentration before the TOP assay. Specifically, this compound oxidizes to form multiple PFCAs, such as PFBA, PFHxA, PFHpA, and PFPeA (Houtz and Sedlak 2012; Martin et al. 2019). In summary, after performance of the TOP assay, 6:2 FTS was not detected, so a near-complete oxidation of this compound, and possibly of unknown precursors, has taken place. Because of this, some PFCAs suffered qualitatively detectable low increases in terms of molar concentration. However, and as has been stated in a previous study, samples with lower total precursor molar concentrations, as is the case of the present research, have significant associated uncertainties with PFCA molar concentrations (Pickard et al. 2022). Thus, the TOP assay performed, reinforced by the results of the previous targeted analysis, provides qualitative information on the presence of precursors in water samples at low concentrations, < 2.5 ng/L, such as 6:2 FTS.
Regarding bottled water, no target precursors included in the method have been detected. Moreover, the TOP assay did not reveal qualitative increases in terms of PFCAs concentration, confirming the absence of other precursors in these water samples.
Exposure and human health risk assessment for PFAS
The estimated weekly intake for the sum of PFOA, PFNA, PFHxS, and PFOS has been determined for two age groups. As shown in Table 6, the exposure for adults, children, and infants suggests that drinking water consumption is responsible for no more than 10%, 21%, and 46% of TWI, respectively. For bottled water, the EWI are below the TWI threshold by a factor percentage of 7%, 21%, and 32% respectively. So, water ingestion of the analyzed samples should not pose any risk to the population. The presented results are in line with those reported by Jurikova et al. (2022), where the evaluation of the ingestion of Czech drinking water samples containing PFAS showed that the recommended guidelines were not exceeded.
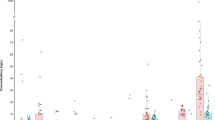
The potential health risks associated with drinking water ingestion containing PFOA, PFNA, PFHxS, and PFOS has been evaluated using hazardous quotients. As shown in Fig. 3, overall, considering DWTP water consumption, HQ values fell within the range of 0.1–1, indicating a low hazard for all age groups. For bottled water, the HQ for adults was < 0.1, representing no hazard. However, for children and infants, the HQ indicated low hazard. It can be concluded that the population exposed to PFAS through ingestion of the mentioned waters does not exceed the current EFSA intake recommendation, suggesting that it may not represent a potential health risk.

Calculated HQ values for average concentration of the sum of PFOA, PFNA, PFHxS, and PFOS. Calculations were performed for different age groups derived from ingestion drinking water from l’Ampolla DWTP and Spanish bottled water
Finally, a worst-case scenario considering the individual parametrical values for PFOA, PFNA, PFHxS, and PFOS stated in the Spanish Royal Decree 3/2023, set at 70 ng/L for each compound, is contemplated to assess if these stablished values might cause adverse effects on human health for each group studied or are restrictive enough. As observed in Table 7, drinking water containing a total concentration of 280 ng/L considerably exceeds the TWI value set by EFSA in each age group, and the related HQs were > 10, indicating that the ingestion of water containing this PFAS concentration may pose a high hazard to human health. This is because the parametrical values stablished in the Spanish or European drinking water directive have no direct connection to EFSAs TWI value for the mentioned fourth PFAS, unlike PFASs levels in food. In this case, it is recommended to revise and stablish new limit values for these compounds, as done in Denmark with a value of 2 ng/L for the sum of these four PFAS (which is derived from the EFSA TWI safe drinking water threshold) in order to ensure sufficient protection for vulnerable groups, avoiding negative effects (Johansson and Hermann 2023).
In view of the obtained results, it is advisable to control the presence of these substances in drinking water, but attention should also be paid to reducing the exposure of the population to PFAS, especially through diet and other routes of exposure, like indoor air inhalation, dust ingestion, and dermal absorption (Domingo and Nadal 2019; Poothong et al. 2020; DeLuca et al. 2022; Ragnarsdóttir et al. 2022).
Conclusions
In this study, an analytical method based on off-line SPE and HPLC–MS/MS has been implemented for the determination of 26 targeted PFAS in Ebro River water, water from pre- and post-ozonation from l’Ampolla DWTP, drinking water and bottled water. Moreover, the TOP assay has been used to assess PFAS precursors. The current methodology employs isotopically labelled compounds as surrogates to compensate SPE recoveries and matrix effect. Although it was impossible to achieve the same labelled compound for each targeted PFAS, the analogues chosen performed, in general, good quantifications. LOQs are in the range of 0.25–5 ng/L, which are optimal for providing information on PFAS content in Spanish drinking water as they are in line with the current Spanish regulation (RD 3/2023). The method was validated and is capable of determining 26 PFAS compounds in different water samples. Firstly, samples from l’Ampolla DWTP were analyzed, presenting low PFAS concentrations (12–16 ng/L). Only 8 targeted compounds were quantified > LOQ. Due to the extremely low concentration of found PFAS, this makes them unlikely compounds to observe a reduction through the treatment processes of the DWTP. As regards bottled water, only one of ten commercial brands analyzed present PFAS (ng/L). The presence of 6:2 FTS (< LOQ) precursors in the mentioned samples has been determined by performing a TOP assay and reveals the absence of any other PFAS precursor in the analyzed sample. Finally, the results obtained were used for estimating health exposure and human health risk, concluding that there is no risk from consuming DWTP drinking water or bottled water.
Data Availability
All data supporting the findings of this study is available from the corresponding author on reasonable request.
References
Al Amin M, Sobhani Z, Liu Y et al (2020) Recent advances in the analysis of per- and polyfluoroalkyl substances (PFAS)—a review. Environ Technol Innov 19:100879. https://doi.org/10.1016/j.eti.2020.100879
Ateia M, Chiang D, Cashman M, Acheson C (2023) Total oxidizable precursor (TOP) Assay─best practices, capabilities and limitations for PFAS site investigation and remediation. Environ Sci Technol Lett 10:2920–3301. https://doi.org/10.1021/acs.estlett.3c00061
Borrull J, Colom A, Fabregas J et al (2020) A liquid chromatography tandem mass spectrometry method for determining 18 per- and polyfluoroalkyl substances in source and treated drinking water. J Chromatogr A 1629:461485. https://doi.org/10.1016/j.chroma.2020.461485
Castiglioni S, Valsecchi S, Polesello S et al (2015) Sources and fate of perfluorinated compounds in the aqueous environment and in drinking water of a highly urbanized and industrialized area in Italy. J Hazard Mater 282:51–60. https://doi.org/10.1016/j.jhazmat.2014.06.007
Chen R, Li G, He Y et al (2021) Field study on the transportation characteristics of PFASs from water source to tap water. Water Res 198:117162. https://doi.org/10.1016/j.watres.2021.117162
Ciofi L, Renai L, Rossini D et al (2018) Applicability of the direct injection liquid chromatographic tandem mass spectrometric analytical approach to the sub-ng L−1 determination of perfluoro-alkyl acids in waste, surface, ground and drinking water samples. Talanta 176:412–421. https://doi.org/10.1016/J.TALANTA.2017.08.052
Coggan TL, Anumol T, Pyke J et al (2019) A single analytical method for the determination of 53 legacy and emerging per- and polyfluoroalkyl substances (PFAS) in aqueous matrices. Anal Bioanal Chem 411:3507–3520. https://doi.org/10.1007/s00216-019-01829-8
DeLuca NM, Minucci JM, Mullikin A et al (2022) Human exposure pathways to poly- and perfluoroalkyl substances (PFAS) from indoor media: a systematic review. Environ Int 162:107149. https://doi.org/10.1016/j.envint.2022.107149
Domingo JL, Nadal M (2019) Human exposure to per- and polyfluoroalkyl substances (PFAS) through drinking water: a review of the recent scientific literature. Environ Res 177:108648. https://doi.org/10.1016/j.envres.2019.108648
Dvorakova D, Jurikova M, Svobodova V et al (2023) Complex monitoring of perfluoroalkyl substances (PFAS) from tap drinking water in the Czech Republic. Water Res 247:120764. https://doi.org/10.1016/j.watres.2023.120764
European Environment Agency (2022) Emerging chemical risks in Europe — ‘PFAS’. https://www.eea.europa.eu/publications/emerging-chemical-risks-in-europe/emerging-chemical-risks-in-europe. Accessed 28 Feb 2023
Glüge J, Scheringer M, Cousins IT et al (2020) An overview of the uses of per- and polyfluoroalkyl substances (PFAS). Environ Sci Process Impacts 22:2345–2373. https://doi.org/10.1039/d0em00291g
Houtz EF, Sedlak DL (2012) Oxidative conversion as a means of detecting precursors to perfluoroalkyl acids in urban runoff. Environ Sci Technol 46:9342–9349. https://doi.org/10.1021/es302274g
Hunt K, Hindle R, Anumol T (2018) Extraction of per / polyfluoroalkyl substances in water using agilent offline solid phase extraction. In: Appl Note. https://www.agilent.com/cs/library/applications/application-infinity-lc-6470-triple-quadrapole-jet-stream-esi-5994-0250en-agilent.pdf. Accessed 5 May 2023
Johansson S, Hermann C (2023) Toxic tide rising:time to takle PFAS. National approaches to address PFAS in drinking water across Europe
Jurikova M, Dvorakova D, Pulkrabova J (2022) The occurrence of perfluoroalkyl substances (PFAS) in drinking water in the Czech Republic: a pilot study. Environ Sci Pollut Res 29:60341–60353. https://doi.org/10.1007/s11356-022-20156-7
Kaboré HA, Vo Duy S, Munoz G et al (2018) Worldwide drinking water occurrence and levels of newly-identified perfluoroalkyl and polyfluoroalkyl substances. Sci Total Environ 616–617:1089–1100. https://doi.org/10.1016/J.SCITOTENV.2017.10.210
Kaiser AM, Saracevic E, Schaar HP et al (2021) Ozone as oxidizing agent for the total oxidizable precursor (TOP) assay and as a preceding step for activated carbon treatments concerning per- and polyfluoroalkyl substance removal. J Environ Manag 300:113692. https://doi.org/10.1016/j.jenvman.2021.113692
Kim KY, Ekpe OD, Lee HJ, Oh JE (2020) Perfluoroalkyl substances and pharmaceuticals removal in full-scale drinking water treatment plants. J Hazard Mater 400:123235. https://doi.org/10.1016/j.jhazmat.2020.123235
Lasier PJ, Washington JW, Hassan SM, Jenkins TM (2011) Perfluorinated chemicals in surface waters and sediments from northwest Georgia, USA, and their bioaccumulation in Lumbriculus variegatus. Environ Toxicol Chem 30:2194–2201. https://doi.org/10.1002/etc.622
Lee YM, Lee JY, Kim MK et al (2020) Concentration and distribution of per- and polyfluoroalkyl substances (PFAS) in the Asan Lake area of South Korea. J Hazard Mater 381:120909. https://doi.org/10.1016/j.jhazmat.2019.120909
Liu S, Junaid M, Zhong W et al (2020) A sensitive method for simultaneous determination of 12 classes of per- and polyfluoroalkyl substances (PFASs) in groundwater by ultrahigh performance liquid chromatography coupled with quadrupole orbitrap high resolution mass spectrometry. Chemosphere 251:126327. https://doi.org/10.1016/j.chemosphere.2020.126327
Martin D, Munoz G, Mejia-Avendaño S et al (2019) Zwitterionic, cationic, and anionic perfluoroalkyl and polyfluoroalkyl substances integrated into total oxidizable precursor assay of contaminated groundwater. Talanta 195:533–542. https://doi.org/10.1016/j.talanta.2018.11.093
McMahon PB, Tokranov AK, Bexfield LM et al (2022) Perfluoroalkyl and polyfluoroalkyl substances in groundwater used as a source of drinking water in the eastern United States. Environ Sci Technol 56:2279–2288. https://doi.org/10.1021/ACS.EST.1C04795/SUPPL_FILE/ES1C04795_SI_001.PDF
Ministerio de la Presidencia R con las C y MD (2023) Royal Decree 3/2023, of January 10, which establishes the technical-sanitary criteria for the quality of drinking water, its control and supply. Boletín Oficial del estado, núm 9, de 11/01/2023
Mottaleb MA, Ding QX, Pennell KG et al (2021) Direct injection analysis of per and polyfluoroalkyl substances in surface and drinking water by sample filtration and liquid chromatography-tandem mass spectrometry. J Chromatogr A 1653:462426. https://doi.org/10.1016/j.chroma.2021.462426
Mussabek D, Söderman A, Imura T et al (2023) PFAS in the drinking water source: analysis of the contamination levels, origin and emission rates. Water 15(1):137. https://doi.org/10.3390/w15010137
Nikiforov VA (2021) Hydrolysis of FTOH precursors, a simple method to account for some of the unknown PFAS. Chemosphere 276:130044. https://doi.org/10.1016/j.chemosphere.2021.130044
Nolan A, Grimison C, Iavetz R et al (2019) Improving measurement reliability of the PFAS TOP assay. In: Proceedings of the 8th international contaminated site remediation conference, Australia, p 633
Organization for Economic Cooperation and Development (OECD) (2023) Portal on per and poly fluorinated chemicals. https://www.oecd.org/chemicalsafety/portal-perfluorinated-chemicals/countryinformation/european-union.htm. Accessed 1 Mar 2023
Pickard HM, Ruyle BJ, Thackray CP et al (2022) PFAS and precursor bioaccumulation in freshwater recreational fish: implications for fish advisories. Environ Sci Technol 56:15573–15583. https://doi.org/10.1021/acs.est.2c03734
Pilli S, Pandey AK, Pandey V et al (2021) Detection and removal of poly and perfluoroalkyl polluting substances for sustainable environment. J Environ Manag 297:113336. https://doi.org/10.1016/j.jenvman.2021.113336
Pizzurro DM, Seeley M, Kerper LE, Beck BD (2019) Interspecies differences in perfluoroalkyl substances (PFAS)toxicokinetics and application to health-based criteria. Regul Toxicol Pharmacol 106:239–250. https://doi.org/10.1016/j.yrtph.2019.05.008
Poothong S, Papadopoulou E, Padilla-Sánchez JA et al (2020) Multiple pathways of human exposure to poly- and perfluoroalkyl substances (PFASs): from external exposure to human blood. Environ Int 134:105244. https://doi.org/10.1016/j.envint.2019.105244
Ragnarsdóttir O, Abdallah MAE, Harrad S (2022) Dermal uptake: an important pathway of human exposure to perfluoroalkyl substances? Environ Pollut 307:119478. https://doi.org/10.1016/j.envpol.2022.119478
Rahman MF, Peldszus S, Anderson WB (2014) Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: a review. Water Res 50:318–340. https://doi.org/10.1016/j.watres.2013.10.045
Schrenk D, Bignami M, Bodin L et al (2020) Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J 18(9):e06223. https://doi.org/10.2903/j.efsa.2020.6223
Schultz MM, Barofsky DF, Field JA (2006) Quantitative determination of fluorinated alkyl substances by large-volume-injection liquid chromatography tandem mass spectrometry - characterization of municipal wastewaters. Environ Sci Technol 40:289–295. https://doi.org/10.1021/es051381p
Schymanski EL, Zhang J, Thiessen PA et al (2023) Per- and polyfluoroalkyl substances (PFAS) in PubChem: 7 million and growing. Environ Sci Technol 57:16918–16928. https://doi.org/10.1021/acs.est.3c04855
Sun M, Arevalo E, Strynar M et al (2016) Legacy and emerging perfluoroalkyl substances are important drinking water contaminants in the Cape Fear River watershed of North Carolina. Environ Sci Technol Lett 3:415–419. https://doi.org/10.1021/acs.estlett.6b00398
Takagi S, Adachi F, Miyano K et al (2011) Fate of Perfluorooctanesulfonate and perfluorooctanoate in drinking water treatment processes. Water Res 45:3925–3932. https://doi.org/10.1016/j.watres.2011.04.052
Teymoorian T, Munoz G, Vo Duy S et al (2022) Tracking PFAS in drinking water: a review of analytical methods and worldwide occurrence trends in tap water and bottled water. ACS ES T Water. https://doi.org/10.1021/acsestwater.2c00387
The Interestate Technology and Regulatory Council (2023) PFAS Technical and Regulatory Guidance Document andFact Sheets PFAS - 1: 2.5 PFAS Uses. https://pfas-1.itrcweb.org/2-5-pfas-uses/
Thomas R, Jenkins K, Landale B et al (2020) Evaluation of PFAS treatment technology: alkaline ozonation. Remediation 30:27–37. https://doi.org/10.1002/rem.21654
United States environmental protection agency (2018) Data review and validation guidelines for perfluoroalkyl substances (PFASs) analyzed using EPA method 537
Verma S, Mezgebe B, Sahle-Demessie E, Nadagouda MN (2021a) Photooxidative decomposition and defluorination of perfluorooctanoic acid (PFOA) using an innovative technology of UV–vis/ZnxCu1-xFe2O4/oxalic acid. Chemosphere 280:1–11. https://doi.org/10.1016/j.chemosphere.2021.130660
Verma S, Varma RS, Nadagouda MN (2021b) Remediation and mineralization processes for per- and polyfluoroalkyl substances (PFAS) in water: a review. Sci Total Environ 794:148987. https://doi.org/10.1016/j.scitotenv.2021.148987
Wang Z, Lin T, Chen W (2020) Occurrence and removal of microplastics in an advanced drinking water treatment plant (ADWTP). Sci Total Environ 700:134520. https://doi.org/10.1016/j.scitotenv.2019.134520
World Health Organization (2022) Guidelines for drinking-water quality: 4th edn. incorporating the first addendum and second addenda, Geneva
Zhong T, Lin T, Zhang X et al (2023) Impact of biological activated carbon filtration and backwashing on the behaviour of PFASs in drinking water treatment plants. J Hazard Mater 446:130641. https://doi.org/10.1016/j.jhazmat.2022.130641
Acknowledgements
The authors are grateful to the Consorci d’Aigües de Tarragona (CAT) for their invaluable collaboration.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Joana Martínez and Massimo Picardo. The first draft of the manuscript was written by Joana Martínez and Massimo Picardo. Alejandra Peñalver, Josepa Fabregas, Carme Aguilar, and Francesc Borrull commented on previous versions of the manuscript. Joana Martínez, Massimo Picardo, Alejandra Peñalver, Josepa Fabregas, Carme Aguilar, and Francesc Borrull read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The authors declare that they have read and approved the manuscript and agree that the work is ready for submission.
Consent for publication
The authors gave consent for the publications of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Roland Peter Kallenborn
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
- An off-line SPE HPLC–MS/MS method was validated for PFAS determination in surface, DWTP, and bottled water samples.
- Twenty-six PFAS compounds’ occurrence and distribution were assessed in l’Ampolla DWTP.
- Of 26 PFAS compounds, 8 targeted compounds were quantified > LOQ in DWTP water samples.
- Total oxidizable precursor assay was conducted on all samples, revealing the presence of only 6:2 FTS.
- The risk assessment concluded that there is no risk to the population from consuming DWTP drinking water or bottled water.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martínez, J., Picardo, M., Peñalver, A. et al. Occurrence and health risk assessment of PFAS and possible precursors: a case study in a drinking water treatment plant and bottled water (south Catalonia, Spain). Environ Sci Pollut Res (2024). https://doi.org/10.1007/s11356-024-34805-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11356-024-34805-6