
Abstract
Purpose
PARP inhibitor (PARPi) therapy is approved for patients with metastatic castration-resistant prostate cancer (mCRPC) and homologous recombination repair (HRR) genomic aberrations. However, only a fraction of patients with BRCA1/2 mutations respond to PARPi therapy. In this pilot study, we assess PARP-1 expression in prostate cancer patients with and without HRR genomic alternations using a novel PARP-based imaging agent.
Procedures
Nine advanced prostate cancer patients were studied with PET/CT and [18F]FluorThanatrace (FTT), an analogue of the PARPi rucaparib. Images were analyzed using maximum standardized uptake values (SUVmax). PARP expression was assessed by immunohistochemistry (IHC) when feasible (n = 4).
Results
We found great variability in FTT uptake (SUVmax range: 2.3–15.4). Patients with HRR mutations had a significantly higher SUVmax (p = 0.0379) than patients with non-HRR mutations although there was an overlap in FTT uptake between groups. Three patients without HRR and one with HRR mutations had similarly high PARP1 IHC expression.
Conclusions
FTT-PET/CT may serve as an alternate biomarker for PARP1 expression and a potential method for PARPi treatment selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostate cancer (PC) is one of the most common malignancies in men. However, despite multiple new therapeutic options, the overall survival for metastatic prostate cancer (mPC) has only improved by about 7 months over the last decade [1]. DNA repair pathways, including homologous recombination repair (HRR), are critical to maintaining cellular integrity. Double-strand DNA breaks can be repaired via either homologous recombination or non-homologous end-joining (NHEJ) [2]. Multiple genes, importantly BRCA1 and BRCA2, play a key role in HRR. Furthermore, poly(ADP-ribose) polymerase 1 (PARP-1) is required for single-strand DNA break repair [3, 4]. PARP-1 binds to DNA at sites of DNA breaks and recruits poly(ADP-ribose) (PAR) to facilitate DNA repair [5]. PARP inhibitors (PARPi) impair this process of DNA repair via synthetic lethality and other mechanisms such as PARP trapping on DNA, leading to cell death [6, 7]. However, the frequency of HRR genomic aberrations in PC is relatively low, and potentially actionable somatic HRR mutations are present only in about 20% of mPC patients [8,9,10]. Nonetheless, a randomized clinical trial demonstrated improved progression-free and overall survival with the PARPi olaparib versus those who received androgen receptor targeted therapy in mCRPC patients with BRCA1/2 or ATM mutations [11, 12]. Thus, novel biomarkers beyond HRR mutations are needed to identify additional PC patients who may respond to PARPi.
Several PET radiotracers to image PARP expression have recently been developed [13]. [18F]FluorThanatrace (FTT), and 18F-PARPi bind to PARP1, are derived from rucaparib and olaparib, respectively [13], and are currently being evaluated in breast and ovarian cancers [14, 15]. FTT has been developed at our institution, and we have previously reported that FTT has an optimal biodistribution in humans, and the primary route of excretion is through hepatobiliary system [16]. The normal organs with the highest uptake were the pancreas, spleen, and liver. The objective of the current pilot study was to evaluate the dynamic range of PARP1 expression in prostate cancer patients with and without HRR genomic alternations utilizing FTT-PET.
Materials and Methods
Study Design and Participants
This study was part of a prospective, single-site phase 0 clinical trial to demonstrate the feasibility of imaging PARP1 expression with FTT-PET/CT in cancer patients (NCT02469129, IND 124,116). The study was approved by all institutional regulatory committees and performed according to the Declaration of Helsinki and the rules of Good Clinical Practice guidelines. All subjects gave written informed consent for participation. Trial eligibility for this investigational imaging study was broad; patients with any type of cancer planned to undergo surgical resection/biopsy, or who were planned to receive systemic therapy, were eligible. Subjects were required to have measurable disease per RECIST 1.1 on CT or other standard radiological imaging. Measurable disease was defined as one or more tumor sites measuring > 1 cm in the shortest transaxial diameter. Subjects with disease < 1 cm were ineligible. Patients who had claustrophobia or who were unable to lie in the PET scanner were also excluded. Vital signs and laboratory evaluations (CBC, CMP) were performed before and after completion of FTT-PET/CT for assessment of any clinically significant change that may have been related to FTT administration. Subjects also were closely monitored during the study for any symptoms that may have been related to FTT injection.
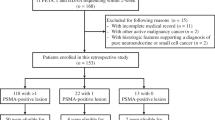
For the present study, patients were consented under the NCT02469129 protocol and were recruited from advanced PC patients in our clinical practice who were either treatment naïve or were scheduled to start a new treatment regimen (Fig. 1). Four patients had metastatic hormone-sensitive prostate cancer (mHSPC); two patients had de novo metastatic disease, and two had recurrent metastatic disease. The remaining 5 patients had metastatic castration-resistant disease (mCRPC) (Table 1). All patients underwent standard of care bone scintigraphy and computed tomography (CT) at baseline prior to enrollment for measurable disease assessment prior study enrollment. All patients also underwent [18F]fluorodeoxyglucose (FDG)-PET/CT) before or after FTT-PET/CT (mean interval 3.6 days). Subjects were encouraged to undergo follow-up imaging after therapy (minimum of 3 months after therapy), but in our cohort, only three patients (#3, 6 and 8) underwent FTT-PET/CT after therapy (Table 1).

Study schema. mHSPC = metastatic hormone sensitive prostate cancer, mCRPC = metastatic castration resistant prostate cancer.
Genomic Analysis
Baseline metastatic biopsy was not required for study entry, and 7 of 9 patients had metastatic tissue available for genomic analysis. Standard of care next-generation sequencing (NGS) was performed utilizing the Tempus xT 648 platform with matched normal blood analysis for germline testing (Tempus Labs; Chicago, IL, USA, https://www.tempus.com; Table 1). Two of the 9 patients did not have metastatic tissue available and thus underwent peripheral blood analysis for germline genetic analysis with the Ambry and Myriad platforms, respectively. One patient also underwent additional ctDNA testing with Guardant 360 in addition to tissue NGS at a separate timepoint prior to receiving care at our institution (Table 1). Four of the 9 patients had sufficient tissue for PARP1 expression analysis via immunohistochemistry (IHC). For this study, we classified BRCA1/2 and ATM mutations, the genomic aberrations most associated with response to PARPi, as “MUT,” and all other mutations as “WT”.
Imaging and Analysis
FTT was prepared as previously described in our cyclotron GMP facility using the GE FXNPro module under current good manufacturing practice (cGMP) guidelines [14]. Briefly, FTT was synthesized under cGMP conditions using a GE FXN Pro reaction module via nucleophilic substitution of a tosylate precursor with a solution of pre-dried [18F]KF/K2CO3/Kryptofix2.2.2 in DMSO at 105 °C. The crude FTT was then purified via a reversed phase HPLC (Zorbax SB-C18, 250 × 9.4 mm, mobile phase 15% acetonitrile (0.1% TFA) in water). The isolated peak was diluted in 40 mL of sterile water for injection (SWI), trapped on an HLB cartridge, eluted from the cartridge using 0.9 mL of ethanol (200 Proof), and formulated in normal saline to provide a 10% ethanol/saline solution. This was sterile filtered through a 0.2-micron Millex-GV filter directly into a sterile final product vial. The final product was analyzed by HPLC, GC, pH, filter membrane integrity test, appearance, color, radiochemical identity, bacterial endotoxin test, and sterility was confirmed as a post release test. All release criteria were met for all injections [14, 16]. The final product meets the releasing criteria approved by FDA.
Patients underwent a 60-min dynamic study immediately following intravenous administration of FTT (median 370 MBq, range 336.7–388.5) over the largest and/or the previously biopsied lesion. Emission imaging were then obtained extending from the skull base to the upper thighs. FTT-PET images were evaluated for areas of abnormal tracer uptake in comparison with anatomical, bone scintigraphy and FDG-PET/CT images using tumor maximum standardized uptake value (SUVmax). For each of the 9 patients, the SUVs of 5 lesions (typically, the largest) were determined, and a total of 45 different disease sites were evaluated.
Immunohistochemistry (IHC) Staining
Biopsy samples were analyzed for PARP1 using an immunohistochemistry (IHC) staining protocol previously described by our group with modifications [17]. Briefly, formalin-fixed paraffin-embedded tissue Sects. (5 µm) were deparaffinized, rehydrated, and subjected to antigen retrieval (citrate buffer, pH 6.0). The sections were blocked (10% normal goat serum), and immunostained with PARP1 (46D11) rabbit antibody (1:300; #9532, Cell Signaling Technologies, USA) followed by ImmPRESS goat anti-rabbit secondary antibody (Vector Laboratories, USA). Diaminobenzidine (Agilent Dako, USA) was used as a chromogen, and sections were counterstained with hematoxylin, dehydrated, cleared, and mounted with Cytoseal XYL (Thermo Scientific, USA). Images were captured using a BX51 microscope (Olympus, Japan). Scoring was performed by an experienced genitourinary pathologist blinded to the clinicopathologic data of the patients. Scoring was adapted from the Allred model of breast cancer estrogen receptors/progesterone receptor staining, whereby an IHC score is calculated as the sum of the positive proportion (0 = 0% positive tumor cells; 1 = < 1%; 2 = 1–10%; 3 = 11–33%; 4 = 34–66%; 5 = 67–100%) and the staining intensity (0 = no staining; 1 = weak; 2 = moderate; 3 = strong) for a possible total score of 8. PARP1 was considered positive if the IHC score was greater than 4.
Statistical Analysis
Demographic and clinical characteristics were summarized by descriptive statistics. The linear mixed effects model for repeated measures was applied, separately to FTT SUVmax in raw and log scales, to model the effect of gene mutation while accounting for the repeated measures of tumor SUVmax for the same patient. The model residuals were examined for goodness of fit, which showed better model fitting using the SUVmax in log scale. The least square mean for MUT and WT was reported with associated standard error and Wald test P value testing the estimated mean against 0. The least square mean difference between MUT (n = 5) and WT (n = 4) patients was reported with associated standard error, the Wald test p value testing the mean difference against 0, and the type III test p value comparing the model with mutation versus the null model without.
Results
Nine PC patients were enrolled (median age of 67 years, range 56–80). Patient characteristics are summarized in Table 1. From a radiotracer safety perspective, no clinically detectable pharmacological effects of FTT or changes in the results of laboratory studies were observed—that is, patients in this study did not have any appreciable alterations in blood chemistries or vital signs after FTT administration.
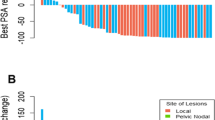
The imaging data were derived from 45 different disease sites: bone (n = 18), liver (n = 2), prostate/prostate bed (n = 6), adrenal gland (n = 1), and lymph nodes (n = 18) (Figs. 1 and 2). As would be expected for advanced prostate cancer, a broad range of organs were involved with metastatic disease: 2 patients had osseous and lymph node metastases, one had osseous and soft tissue, one had osseous and hepatic, 1 had osseous and adrenal gland, 1 had osseous, and 1 had lymph node metastases. The SUV uptake for each of the studied lesions is delineated in Table 2.

The graph is patient ID on the x-axis plotted against FTT SUVmax on the y-axis. Patients are arranged on the x-axis in ascending order of FTT SUVmax. Red dots indicate MUT tumor lesions and cyan triangles indicate WT tumor lesions. The black horizontal line indicates the median SUVmax across all lesions for a given patient. There was a statistically significant difference between the SUVmax of the MUT (n = 5) and WT (n = 4) patients (p = 0.0379). MUT, BRCA1/2, ATM; WT, non-BRCA1/2 or ATM mutations.
The SUVmax for FTT ranged from 2.3 (similar to normal bone) to 15.4 in osseous lesions (Fig. 2). We did not identify a significant difference in SUVmax between osseous (mean and range 8.82; 3.6–14.2) and lymph nodes/soft tissue lesions (7.50; 2.3–15.4, p = 0.6471). After linear mixed effects modeling of FTT SUVmax on a log scale (log scale FTT SUVmax difference mean/SE = 0.4306/0.1685), patients with MUT genomic alterations had a significantly higher (Wald test P = 0.0379) FTT SUVmax than patients with WT alterations (Fig. 2). Patients 3, 9, 7, and 8, all of whom harbored either germline or somatic BRCA2 alterations, had the highest FTT uptake. Although patient 4 (Fig. 3), who harbored an ATM LOF mutation and was categorized as MUT, the uptake for this patient was proportionally lower compared with the BRCA mutant patients. The patients with the lowest SUVmax harbored TP53, RB1, RAD50, and CDK12 mutations. Of note, both patients 3 (BRCA2 copy number loss) and patient 8 (simultaneous BRCA2 germline alteration and somatic mutation) underwent a baseline FTT PET and then initiated treatment with standard of care olaparib. Follow-up FTT-PET/CT was completed at 9 and 10 months (Fig. 4), respectively. At the time of the follow-up imaging, the PSA markedly decreased in patient 3 (88.86 to 0.90 ng/mL) and patient 8 (44.72 to 0.90 ng/mL), correlating with excellent radiographic response.

Prostate adenocarcinoma (#4). Fused FTT-PET/CT images (left) and CT images (right). There are FTT + right perirectal (top row) and right internal iliac lymph nodes (bottom row arrows). The scale bar on PET images: SUVmax = 5.5

Prostate adenocarcinoma (#6). A Fused FTT-PET/CT (left top row) and CT (right: top, bone window; bottom, soft tissue window) images. There is FTT + sternal metastasis (arrows) with slightly different distribution. Additional FTT + mediastinal lymph nodes are noted. B PARP1 IHC score of 8. C Fused FTT-CT sagittal images of before and 4 months after PARPi therapy (#6 non-responder). The post-therapy image demonstrates the resolution of the sternal lesion (arrow) seen on pre-therapy image and appearance of a new sternal lesion just inferior to the initial lesion (arrow). D Fused FTT-CT sagittal images of before and 4 months after PARPi therapy (#8 responder). The post-therapy image demonstrates near-complete resolution of all spine and sternal metastatic disease. The scale bar on PET images: SUVmax = 5.5
Patient 6 had only one site of known disease (sternum) on CT, FDG-PET/CT and bone scintigraphy and several additional sites of FTT avid osseous and lymph node disease, which were suspected to be additional sites of disease. He was initiated on olaparib therapy due to a CDK12 tumor mutation and an ATM mutation on cell-free DNA liquid biopsy. However, the ATM variant allele fraction (VAF) was very low at 0.2%, suggesting a likely subclonal alteration. On post-therapy scan 5 months later, the patient’s PSA increased (4.61 ng/mL to 8.26), correlating with disease progression on FTT-PET/CT and correlative imaging. The mean SUV of these lesions were lower than that in patients # 3 and # 8 (6.5 vs. 7.2 and 13.04, respectively) who responded to PARPi therapy (Fig. 4).
IHC analysis demonstrated high PARP1 expression (score 5 + 3 = 8 in 3 WT patients and 5 + 2 = 7 in 1 MT patient), correlating with high FTT uptake (Table 1, Fig. 4).
Discussion
Despite six FDA-approved treatments for mCRPC, long-term survival outcome in advanced PCa remains poor, and further treatment advances are needed. mCRPC with HRR genomic aberrations have been shown to be sensitive to PARPi [11, 18]. However, based on the PROfound trial, patients with non-BRCA/ATM genomic aberrations (BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L) do not benefit to the same degree as patients with alterations in BRCA1/2 or ATM (response rate to olaparib: 4% vs. 33%, respectively) [19, 20]. These mixed results have also been demonstrated in the TRITON2 trial, wherein patients with deleterious BRCA1/2 alterations had greater response to rucaparib than patients with ATM, CHEK2, and CDK12 mutations [18, 21]. Thus, the role of HRR genomic aberrations in accurately predicting response to PARPi remains unclear. Therefore, it is of critical importance to identify a means for selecting patients likely to respond to PARPi that is not solely dependent on evaluating HRR status. In our study, patients harboring either somatic or germline BRCA2 genomic alterations had the highest FTT uptake, and two of these patients also demonstrated radiographic response on follow-up FTT-PET that correlated with clinical PSA response to PARPi therapy. However, particularly noteworthy is that fact that the patient with a CDK12 mutation who received olaparib did not have a favorable response on FTT-PET. This lack of clinical benefit correlates with the lack of radiographic benefit reported in TRITON2 in CDK12 patients (0 of 10 patients), suggesting that lack of high FTT uptake may be predictive of low likelihood of response to PARPi [22].
Currently, there are several ongoing clinical trials of FTT-PET/CT in breast, ovarian, pancreatic, brain, and prostate cancers. Several studies have shown the specificity of FTT for in vivo quantitative imaging of PARP1 [15, 23]. Similar results have been reported in breast cancer patients where FTT uptake was independent of breast cancer subtypes BRCA pathogenic variant carriers and non-carriers [24]. Our study demonstrates the feasibility of FTT-PET/CT in PC patients as a potential imaging biomarker to augment patient selection appropriate for PARPi, and we found a wide range of FTT uptake independent of HRR mutation status.
Furthermore, one patient with MUT and 3 patients with WT had PARP1 IHC analysis and demonstrated high expression, suggesting that tumor suppressor gene loss may also be associated with PARP1 expression. Alternatively, this may simply be confirming highly variable PARP-1 IHC regardless of BRCA status in patients with ovarian, breast, and prostate cancers, as shown by others [25,26,27]. Thus, an in vivo imaging biomarker such as FTT uptake may serve as a putative biomarker for potential response to PARPi and may be a preferred mode in selection of appropriate patients for PARPi therapy. The main limitation of our study is the small sample size. Additional subjects are needed to fully characterize the utility of the FTT as an imaging biomarker for selection of patients who will respond to PARPi therapy. In addition, the correlation between PARP1 expression and response to therapy independent of established HRR genomic aberrations need to be evaluated.
Conclusion
In the present pilot study, we demonstrated the feasibility of imaging advanced PC patients with FTT-PET/CT. The studied lesions had a broad range of SUV uptake independent of HRR status, but with overall higher uptake in patients harboring BRCA2 mutations. Our preliminary results support further investigation of FTT-PET/CT as a technology to evaluate PARP1 expression in vivo, with potential application as a biomarker for PARPi treatment selection in advanced PC.
References
Francini E, Gray KP, Shaw GK, Evan CP, Hamid AA, Perry CE et al (2019) Impact of new systemic therapies on overall survival of patients with metastatic castration-resistant prostate cancer in a hospital-based registry. Prostate Cancer Prostatic Dis 22(3):420–427
Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5(11):a012740
Helleday T (2011) The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5(4):387–393
Fisher AE, Hochegger H, Takeda S, Caldecott KW (2007) Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol 27(15):5597–5605
Langelier MF, Zandarashvili L, Aguiar PM, Black BE, Pascal JM (2018) NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat Commun 9(1):844
Lord CJ, Ashworth A (2017) PARP inhibitors: synthetic lethality in the clinic. Science 355(6330):1152–1158
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH et al (2012) Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72(21):5588–5599
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H et al (2016) Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 375(5):443–453
Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R, Millis SZ et al (2019) Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol. 3.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM et al (2015) Integrative clinical genomics of advanced prostate cancer. Cell 162(2):454
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S et al (2020) Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 382(22):2091–2102
Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S et al (2020) Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med 383(24):2345–2357
Puentes LN, Makvandi M, Mach RH (2021) Molecular imaging: PARP-1 and beyond. J Nucl Med 62(6):765–770
Zhou D, Chu W, Xu J, Jones LA, Peng X, Li S et al (2014) Synthesis, [(1)(8)F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography. Bioorg Med Chem 22(5):1700–1707
Edmonds CE, Makvandi M, Lieberman BP, Xu K, Zeng C, Li S et al (2016) [(18)F]FluorThanatrace uptake as a marker of PARP1 expression and activity in breast cancer. Am J Nucl Med Mol Imaging 6(1):94–101
Li S, Schmitz A, Lee H, Mach RH (2017) Automation of the radiosynthesis of six different (18)F-labeled radiotracers on the AllinOne. EJNMMI Radiopharm Chem 1(1):15
Huynh TT, Sreekumar S, Mpoy C, Rogers BE (2022) Therapeutic Efficacy of (177)Lu-Labeled A20FMDV2 Peptides Targeting alphanubeta6. Pharmaceuticals (Basel). 15(2)
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R et al (2020) Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol 38(32):3763–3772
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R et al (2020) Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 21(1):162–174
Stopsack KH (2021) Efficacy of PARP Inhibition in metastatic castration-resistant prostate cancer is very different with non-BRCA DNA Repair alterations: reconstructing prespecified endpoints for cohort B from the phase 3 PROfound trial of olaparib. Eur Urol 79(4):442–445
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ et al (2020) Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase 2 TRITON2 study. Clin Cancer Res
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ et al (2020) Non-BRCA DNA Damage repair gene alterations and response to the PARP Inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res 26(11):2487–2496
Michel LS, Dyroff S, Brooks FJ, Spayd KJ, Lim S, Engle JT et al (2017) PET of Poly (ADP-ribose) polymerase activity in cancer: preclinical assessment and first in-human studies. Radiology 282(2):453–463
McDonald ES, Doot RK, Pantel AR, Farwell MD, Mach RH, Maxwell KN et al (2020) Positron emission tomography imaging of poly-(adenosine diphosphate-ribose) polymerase 1 expression in breast cancer: a nonrandomized clinical trial. JAMA Oncol 6(6):921–923
Makvandi M, Pantel A, Schwartz L, Schubert E, Xu K, Hsieh CJ et al (2018) A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J Clin Invest 128(5):2116–2126
Domagala P, Huzarski T, Lubinski J, Gugala K, Domagala W (2011) PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast Cancer Res Treat 127(3):861–869
Pignochino Y, Capozzi F, D’Ambrosio L, Dell’Aglio C, Basirico M, Canta M et al (2017) PARP1 expression drives the synergistic antitumor activity of trabectedin and PARP1 inhibitors in sarcoma preclinical models. Mol Cancer 16(1):86
Funding
FD received funding from the Siteman Cancer Center/Barnes-Jewish Hospital Foundation for the conduct of the study. This work also supported by the Response Assessment and Biostatistics Shared Resource Cores of the Siteman Cancer Center (supported in part by NCI Grant P30 CA91842).
Author information
Authors and Affiliations
Contributions
All authors participated in the writing the manuscript. In addition, FD led and supervised the trial and participated in study design, data analysis, data interpretation, and preparing figures/tables. MAR and RP participated in patient recruitment and data analysis, KLS participated in data analysis, and preparing figures/tables, DLC participated in the study design, JL participated in statistical analysis of the data, SS, CW and BR participated in IHC preparation and analysis, DZ participated in data analysis.
Corresponding author
Ethics declarations
Conflict of Interest
FD, MAR, KIS, JL, SS, CW, BR, RKP, and DZ report no conflicts of interest. DLC is a co-inventor on US Patent WO 2015103526 A1 that covers 18F-FTT, held by Washington University and licensed to Trevarx Biomedical, and thus, they may receive royalty income. No other potential conflicts of interest relevant to this article exist.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dehdashti, F., Reimers, M.A., Shoghi, K.I. et al. Pilot Study: PARP1 Imaging in Advanced Prostate Cancer. Mol Imaging Biol 24, 853–861 (2022). https://doi.org/10.1007/s11307-022-01746-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-022-01746-w