
Abstract
Introduction
Autoimmune disorders such as type 1 diabetes (T1D) are believed to be caused by the interplay between several genetic and environmental factors. Elucidation of the role of environmental factors in metabolic and immune dysfunction leading to autoimmune disease is not yet well characterized.
Objectives
Here we investigated the impact of exposure to a mixture of persistent organic pollutants (POPs) on the metabolome in non-obese diabetic (NOD) mice, an experimental model of T1D. The mixture contained organochlorides, organobromides, and per- and polyfluoroalkyl substances (PFAS).
Methods
Analysis of molecular lipids (lipidomics) and bile acids in serum samples was performed by UPLC-Q-TOF/MS, while polar metabolites were analyzed by GC-Q-TOF/MS.
Results
Experimental exposure to the POP mixture in these mice led to several metabolic changes, which were similar to those previously reported as associated with PFAS exposure, as well as risk of T1D in human studies. This included an increase in the levels of sugar derivatives, triacylglycerols and lithocholic acid, and a decrease in long chain fatty acids and several lipid classes, including phosphatidylcholines, lysophosphatidylcholines and sphingomyelins.
Conclusion
Taken together, our study demonstrates that exposure to POPs results in an altered metabolic signature previously associated with autoimmunity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Type 1 diabetes (T1D) is one of the most common chronic metabolic disorders in children and adolescents, with an estimated worldwide incidence of 128,900 new cases and corresponding prevalence of nearly 1,100,000 existing cases in the under 20 year age group (Patterson et al., 2019). Genetic factors are known to play a significant role in the development of autoimmune disorders, including T1D in humans, potentially in combination with environmental factors (Bach, 2001; Regnell & Lernmark, 2013; Rook, 2012). In recent decades, age at diagnosis of T1D has declined (Group, 2008; Harjutsalo et al., 2013), suggesting that the impact of environmental factors during early development has increased. Factors such as maternal infections or infections in early life, deficiency of specific nutrients during pregnancy and/or early childhood, have been associated with risk of T1D in observational studies (Stene & Gale, 2013). Other proposed risk factors of T1D include alterations in gut microbiota (Chatenoud et al., 2012) and exposure to environmental chemicals during development (Bodin et al., 2015). However, the impact of environmental factors on disease development is not well characterized. While epidemiological studies have identified specific associations between environmental factors and disease risk, causal relationships require also controlled, experimental in vitro and in vivo exposure models.
Environmental chemicals, particularly persistent organic pollutants (POPs) including per- and polyfluoroalkyl substances (PFAS), organochlorine pesticides (OCPs), polychlorinated biphenyls (PCBs) and brominated flame retardants (BFRs), such as hexabromocyclododecane (HBCD) and polybrominated diphenyl ethers (PBDEs), are commonly detected in humans as demonstrated by the European Human BioMonitoring data dashboard (HBM4EU, 2020). Although humans are exposed to complex, man-made chemical mixtures via several sources and routes, knowledge of adverse effects caused by such chemicals is mainly based on studies focusing on a single or few chemicals at a time. However, combined exposure to multiple chemicals may cause synergistic or additive adverse health effects, even if the levels of all substances included in the chemical exposure mixture are below their individual safety thresholds (JRC, 2018).
Humans are exposed to PFAS, PCBs and BFRs through food (Harrad et al., 2004; Haug et al., 2011; Hites et al., 2004; Karrman et al., 2006), via inhalation (Casey et al., 1999; Mandalakis et al., 2008) and indoor dust (Hazrati & Harrad, 2006; Hwang et al., 2008; Wilford et al., 2004), as well as through breastfeeding (Karrman et al., 2007). After intake and absorption, perfluorinated compounds predominantly associate with proteins and occur at the highest concentrations in blood, liver and kidneys (Karrman et al., 2006; Lau, 2012), while the main site of PCB and BFR accumulation is adipose tissue (Mullerova & Kopecky, 2007). During the early development in humans, POPs can pass the placental barrier, reach and deposit in fetal tissues and organs (Li et al., 2020a; Porpora et al., 2013). They are, in general, found in fetal organs at concentrations lower than in maternal serum, but similar to placental levels (Mamsen et al., 2019).
In living organisms, many POPs are known to cause adverse health impacts. For example, PFAS exposure has been associated with endocrine disruption (Lopez-Espinosa et al., 2012), increased cholesterol levels (Li et al., 2020b), altered metabolic processes in fat and liver (Bassler et al., 2019), and has been linked with autoimmune disorders such as celiac disease (Sinisalu et al., 2020) and type 1 diabetes (McGlinchey et al., 2020). Several PCB congeners have been associated with adverse developmental (Mol et al., 2002), endocrine (Cao et al., 2008; Chen et al., 2008; Vasiliu et al., 2006) and immunological (Heilmann et al., 2006; Lee et al., 2007; Park et al., 2008) effects. Adverse health consequences of exposure to BFRs have also been observed, including thyroid disorders (Stapleton et al., 2011; Zota et al., 2011), diabetes (Lee et al., 2010) and neurodevelopmental (Gascon et al., 2011) health effects.
The number of exposure studies investigating autoimmune disease progression in animal models are limited. We have previously reported that exposure to a single PFAS, perfluoroundecanoic acid (PFUnDA), caused a non-monotonic dose–response effect on the immune system in non-obese diabetic (NOD) mice, where low and intermediate exposures resulted in a delay of onset of autoimmune diabetes, while exposure to very high levels accelerated insulitis development (Bodin et al., 2016). Further, exposure of the NOD mice to a POP mixture containing 7 PCBs, 9 OCPs, 7 BFRs and 6 PFAS (Berntsen et al., 2017) resulted in a trend of decreased peritoneal macrophage phagocytosis (Berntsen et al., 2018). Additionally, an early event of insulitis development, namely a reduced number of F4/80-tissue resident macrophages, was also apparent in the histological analysis of pancreas in these mice. We have previously shown that immunotoxic effects of a POP mixture in in vitro studies of both rodent and human blood-derived macrophages were mainly caused by the PFAS in the mixture (Berntsen et al., 2018).
Human studies investigating early life exposure to POPs and their role in T1D development show inconsistent results. A cross-sectional study including 820 patients with T1D showed a significant association between high PFAS levels in blood serum and lower risk of T1D development in children and adults (Conway et al., 2016). Conversely, elevated perfluorooctanesulfonic acid (PFOS) levels were reported in young patients newly diagnosed with T1D (Predieri et al., 2015), while another birth-cohort study found no evidence that fetal and early life exposure to POP (including 14 PFAS) was a significant risk factor for later T1D development (Salo et al., 2019). Other studies suggest that PFAS exposure is associated with hyperglycemia, serum high-density lipoprotein (HDL) cholesterol and increased blood insulin (Lin et al., 2009), may lead to immunotoxicity (Borg et al., 2013), and modulates neonatal serum phospholipids associated with increased risk of T1D (McGlinchey et al., 2020). Overall, the molecular mechanisms underlying the effects of exposure are still not well characterized.
Herein we investigated metabolic alterations in NOD mice following pre- and postnatal exposure to a mixture of persistent organic pollutants, in which the concentrations of POPs were based on estimated daily intake levels reported in the Nordic population (Berntsen et al., 2017). Since PFAS was the most potent chemical group in the POP mixture regarding macrophage phagocytosis (Berntsen et al., 2018), we focused on possible effects of PFAS on the metabolome.
2 Materials and methods
2.1 Exposure mixture
All 29 compounds in the POP mixture (Table 1) were of HPLC/GC–MS grade (at least > 98.5% purity) and underwent a quality check for the absence of any dioxin-like compounds by the suppliers (Chiron AS, Norway; Sigma-Aldrich, Germany; Santa Cruz Biotechnology, Inc., USA).
Three exposure diets control (solvents only), low exposure (5000 × human estimated daily intake (EDI)) and high exposure (100,000 × human EDI), were prepared by dissolution of POPs in corn oil intended for human consumption (Yonca Gida Sanayi A.S., Turkey), which was then incorporated into pelleted feed (TestDiet, USA). The EDI values in low and high exposure diets (ng/kg BW/day in humans) were adjusted to a mean mouse bodyweight of 25 g, assuming 3 g per day feed intake, as earlier described (Berntsen et al., 2017). The total average daily intake of OCPs and PCBs was 1.8 µg and 35.0 µg; of BFRs 0.40 µg and 8.0 µg; and of PFAS 0.14 µg and 2.9 µg in the low and high exposure groups, respectively.
2.2 NOD mice study
All animal experiments were approved by the Norwegian Animal Research Authority and conducted in accordance with the Norwegian laws and regulations for experiments using live animals. The study design was reported previously (Berntsen et al., 2018).
Twenty female and ten male NOD/SHiLtJ mice (Jackson Laboratory, USA) were randomized into exposure groups and used for breeding at 8 and 10 weeks of age. The POP exposure via feed started at mating and continued through gestation and early life period until 12 weeks of age. Due to developing a more invasive and destructive insulitis (Bao et al., 2002; Kelemen, 2004), only female offspring were selected at weaning. At 12 weeks of age, eight female offspring were sacrificed in the control group along with seven female offspring from each of the low and high exposure groups.
Blood serum samples were collected and stored at -80 °C until chemical analyses. Samples were prepared and analyzed by mass spectrometry in randomized order.
2.3 Lipidomic analysis
A previously published and modified Folch procedure (Nygren et al., 2011) was used to extract blood serum samples. Briefly, 10 µL blood serum sample aliquots were extracted with 10 µL 0.9% NaCl and 120 µL 2:1 v/v trichloromethane:methanol (Fisher Scientific, UK) solution containing 2.5 µg/mL internal standards purchased from Avanti Polar Lipids Inc., USA and Larodan AB, Sweden (Supplementary Table A1). Samples were vortexed and kept on ice for 30 min prior to 3 min of centrifugation at 9400 × g. Aliquots of 60 µL bottom layer were transferred to new vials and 60 µL of 2:1 v/v trichloromethane:methanol solution were added to each sample.
Calibration standards at concentration levels of 100, 500, 1000, 1500, 2000 and 2500 ng/mL were prepared in 2:1 v/v trichloromethane:methanol solution using supplied by Avanti Polar Lipids Inc., USA and Larodan AB, Sweden chemicals (Supplementary Table A1).
After the extraction, samples were analyzed utilizing ultra-high performance liquid chromatography quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF/MS). An Agilent Technologies 1290 Infinity II system was used, connected to a 6545 Q-TOF/MS with MassHunter B.06.01 software installed to acquire the data. A multisampler, maintained at 10 °C, a column thermostat at 50 °C and a quaternary solvent manager were used to introduce samples onto 1.7 µm, 2.1 mm × 100 mm Acquity UPLC BEH C18 column (Waters Corporation, USA). Solvent and extraction blanks, pure lipid standard samples representing each class of lipids, pooled serum samples and reference plasma samples SRM1950 (NIST, USA) were used to perform a quality control of the analysis. Relative standard deviations (RSD) of peak areas were within accepted analytical limits and were on average 11.9% in lipid standards and 7.3% in pooled control samples.
After the acquisition, the data processing was performed in MZmine 2.18 (Pluskal et al., 2010) applying following steps and options: (1) crop filtering: m/z 350–1200 and retention time (RT) 2.0–12 min; (2) mass detection: noise level at 750; (3) chromatogram builder: minimum time span – 0.8 min, minimum height 1000, m/z tolerance 0.006 m/z or 10.0 ppm; (4) chromatogram deconvolution utilizing the local minimum search algorithm: chromatographic threshold 70%, minimum RT range 0.05 min, minimum relative height 5%, minimum absolute height – 1200, minimum top-to-edge ratio 1.2, peak duration range 0.08–5.0; (5) isotopic peak grouper with the most intense peak as the representative isotope: m/z tolerance 5.0 ppm, RT tolerance 0.05 min, maximum charge 2; (6) peak filter: minimum data points 12, full width at half maximum (FWHM) 0.0–0.2, tailing factor 0.45–2.22, asymmetry factor 0.40–2.5; (7) join aligner: m/z tolerance 0.009 m/z or 10 ppm with weight of 2, RT tolerance 0.1 min with weight of 1, with no requirement of charge state or ID, no comparison of isotope pattern; (8) peak list row filter: feature present minimum In 10% of samples; (9) gap filling using the “same RT and m/z range” algorithm: m/z tolerance 0.009 m/z or 11 ppm; (10) lipids identification using custom database search: m/z tolerance 0.009 m/z or 10.0 ppm and RT tolerance 0.1 min; (11) peak area normalization based on: internal standards of the same lipid classes for identified lipids (PE(17:0/17:0), SM(d18:1/17:0), Cer(d18:1/17:0), LPC(17:0), TG(17:0/17:0/17:0) and PC(16:0/d30/18:1)) and internal standards with closest RT for the unidentified lipids; (12) calculations of concentrations using previously-described 6-point calibration curves. Furthermore, the final dataset was subjected to further data clean-up steps by removing compounds present at high levels in extraction blanks (median samples/median blanks > 3), as well as removing compounds with relative standard deviation in the pooled samples > 30% and including compounds having a detection rate > 70% after the filtering and clean-up steps. Identification was based on in-house spectral library based on m/z, MS/MS and retention time data.
2.4 Analysis of bile acids and PFAS
Serum samples were analyzed for bile acids (BAs) and PFAS employing modified and previously described method (Salihovic et al., 2020). Samples were randomized and 50 µL serum aliquots were extracted with 100 µL acetonitrile (Fisher Scientific, UK), with addition of 10 µL 200 µg/mL PFAS internal standards (Wellington Labortories, Canada) and 20 µL 440–670 ng/mL BA internal standards (Qmx Laboratories Ltd., UK) in methanol (Supplementary Table A1, see also BA abbreviations).
The samples were centrifuged for 3 min at 9400 × g, organic phase collected and evaporated under gentle nitrogen flow to dryness, and residue was dissolved in 300 µL 2 mM ammonium acetate (Fisher Scientific, UK) in water. After addition of 10 µL 200 µg/mL injection standard containing labelled standards (Wellington Laboratories, see Supplementary Table A1), samples were analyzed on Acquity UPLC instrument coupled to a triple quadrupole mass spectrometer (Waters Corporation, USA). A system was equipped with a perfluorinated compounds (PFC) isolator trap column installed between pump and injector, 1.7 µm 2.1 mm × 100 mm UPLC BEH C18 column and an atmospheric electrospray interface operating in negative ion mode. Mobile phases used were: 2 mM NH4Ac in water (A) and 9:1 methanol:2 mM NH4Ac in methanol (B). Following gradient program at constant 0.3 mL/min flow rate was used to separate compounds of interest: up to 1 min, 99% solvent A; 1–16 min 1% solvent A; 16–17 min 99% solvent A. Data acquisition was performed in a multiple reaction monitoring (MRM) mode. Six points external calibration curve at 0.5–160 ng/mL concentration range was used for the quantification of BAs purchased from Sigma-Aldrich, Germany; Steraloids, USA; Calbiochem, USA; and Fluka Chemie, Switzerland.
2.5 Polar metabolites analysis
Protein precipitation was performed with a previously described procedure (Castillo et al., 2011). Aliquots of 30 µL serum were extracted with 400 µL of MeOH containing 1 µg/mL internal standards from Sigma-Aldrich, Germany (Supplementary Table A1). Samples were then vortexed and kept on ice for 30 min, prior to the centrifugation for 3 min at 9400 × g. Supernatants with a volume of 350 µL were transferred to new vials and evaporated to dryness under nitrogen flow. Two-step derivatization was performed by incubation for 60 min at 45 °C at each step, first with 25µL 20 mg/mL metoxamine (MOX) in pyridine and secondly with 25µL n-methyl-n-trimethylsilyl-trifluoroacetamide (MSTFA). A retention index (RI) standard mixture containing 10 µg/mL n-alkanes in hexane was added before the analysis on Agilent 7890B gas chromatograph coupled to 7200 Q-TOF/MS.
A programmable temperature vaporizer (PTV) injector with an initial temperature 70 °C and heated at 120 °C/min to 300 °C was used to inject 1 µL of sample at 100:1 split ratio. A Zebron ZB-SemiVolatiles column (Phenomenex, USA) with 20 m length, 0.18 mm i.d. and 0.18 µm film thickness and 1.2 mL/min helium (Linde Gas AB, Sweden) were used. Oven temperature program was as follows: after 5 min at initial 50 °C, temperature was first raised at 20 °C/min to 270 °C, and then to 300 °C at 40 °C/min with 5 min hold time of the final temperature and 2.4 ml/min helium flow to condition the column. Electron impact (EI) source with 3 min solvent delay, set to 250 °C, 70 eV electron energy and 35 µA emission was used to ionize compounds. Data was acquired by MassHunter software applying mass range of 55–650 amu, 5 spectra/s acquisition rate, 150 °C quadrupole temperature and 1.5 mL/min nitrogen collision gas flow.
The prepared six-point external calibration curve at 0.1–80 µg/mL contained standards purchased from Sigma-Aldrich (Supplementary Table A1). Extraction blanks, pooled aliquots of samples, NIST SRM 1957 certified serum and pooled in-house samples were analyzed to control quality of the performed analysis. Analysis reproducibility was within accepted analytical methods and RSD values of metabolite concentrations in pooled NOD mouse serum samples were lower than 8%. Identification was based on comparison with authentic standards (level 1 identification) or comparison with spectral libraries (NIST2017), using also information on retention index data.
2.6 Statistical analyses
Only metabolites detected in more than 70% of samples were selected. All datasets were then preprocessed in three steps prior to the statistical analyses. At first, values below detection limits were imputed as half the minimum-detected concentrations of the corresponding compound. Secondly, all concentrations were base-2 logarithmically transformed. Finally, all log2-transformed variables were autoscaled, i.e., normalized to zero mean and unit variance.
Analysis of variance (ANOVA) and Tukey’s honest significant differences (HSD) test were generated using Microsoft Excel (Microsoft Corporation, WA, USA). Boxplots were plotted in MATLAB R2018b software (Mathworks Inc., MA, USA). The R statistical programming language and corrplot R package ver.084 were used to calculate and visualize correlations between metabolites and PFAS in NOD mouse blood serum. Principal component analysis (PCA), heatmap analysis and pathway analysis were performed using Metaboanalyst ver 5.0 web-based metabolomics data analysis tool (Xia & Wishart, 2011). The Venn diagram was plotted using the jvenn plug-in (Bardou et al., 2014).
3 Results
3.1 Metabolic profiles of NOD mice
A total 251 metabolites were identified and quantified in 22 NOD mouse blood serum samples (178 lipids, 52 polar metabolites and 21 bile acids). The major lipid classes analyzed were cholesteryl esters (CEs), ceramides (Cers), lysophosphatidylcholines (LPCs), phosphatidylcholines (PCs), phosphatidylethanolamines (PEs), sphingomyelins (SMs) and triglycerides (TGs). Polar metabolites included amino acids, free fatty acids, hydroxy acids and metabolites from central carbon metabolism. One-way ANOVA and Tukey’s HSD tests for post-hoc analysis indicated dysregulation of 14 metabolites in low exposure and 120 metabolites in high exposure NOD mouse groups (Supplementary Table A2).
3.2 PFAS exposure associated with changes in metabolome
PFAS levels in the exposed mice (Table 2) varied from low ppb to ppm levels, with low levels of PFAS detected also in the control mice.
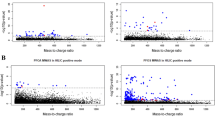
ANOVA with post-hoc Tukey’s HSD tests showed that several bile acids were dysregulated in the exposed groups. Litocholic acid (LCA) was upregulated 2.1-fold (p < 0.01) in the low exposure and 5.9-fold (p < 0.001) in the high exposure versus the control group. In contrast, hyodeoxycholic acid (HDCA) was 4.3-fold downregulated in the high exposure group compared to the controls, while 11 BAs were significantly downregulated in the high versus the low exposure group (Supplementary Table A2). Spearman’s correlation analysis revealed that two bile acids, namely taurodeoxycholic acid TDCA (p < 0.01) and glycodeoxycholic acid GDCA (p < 0.05), were negatively correlated, while LCA (p < 0.001) was positively correlated with PFAS in NOD mouse blood serum samples (Fig. 1). In addition, most of BAs showed tendency of negative correlation to PFAS.

Spearman’s correlations between PFAS and lipid classes (A), bile acids (B) polar metabolites (C), including values from all 22 NOD mouse blood serum samples. Significant correlations marked with *p < 0.05, **p < 0.01 and ***p < 0.001. Positive correlation showed in blue and negative in red
In lipids, the most prominent exposure-induced changes were observed among the LPC, PC and SM lipid classes (Fig. 1). Spearman’s correlation analysis showed that LPC 16:0e (p < 0.001), LPC 16:0p (p < 0.001), LPC 18:2 (p < 0.05), LPC 20:4 (p < 0.05), LPC 22:5 (p < 0.001) and LPC 22:6 (p < 0.05) were inversely, while LPC 20:3 (p < 0.01) was positively correlated with PFAS concentrations. Out of 67 PCs, 46 were negatively and two positively associated with PFAS in mouse serum. All except one of fourteen SMs had inverse correlation with PFAS. Additionally, four TGs correlated with PFAS exposure.
Out of 52 polar metabolites, three, 19 and 16 metabolites showed statistically-significant changes (p < 0.05) in low exposure vs. control, high exposure vs. control and high exposure vs. low exposure groups respectively (Supplementary Table A2).
Correlation between PFAS and polar metabolites, such as amino acids, free fatty acids, short chain fatty acids showed inverse correlation (Fig. 1) of serine (p < 0.05), threonine (p < 0.01), aspartic acid (p < 0.01), glycerol-3-phosphate (p < 0.001), palmitic acid (p < 0.01), linoleic acid (p < 0.05), stearic acid (p < 0.05), arachidonic acid (p < 0.01), cholesterol (p < 0.001) and tentatively identified 1,5-anhydrohexitol (p < 0.001). 3-hydroxybutyric acid (p < 0.001), fumaric acid (p < 0.05), methionine (p < 0.05), glutamic acid (p < 0.001) and tentatively-identified gluconic acid (p < 0.001), uric acid (p < 0.01) and glycerol monostearate (p < 0.05) were positively correlated with PFAS levels.
Principal component analysis (PCA) showed that the metabolic profile in the high exposure group was clearly different to that of the control and low exposure group (Fig. 2A). With the first three principal components (PC) included, the explained cumulative variance of the model was 62.1%. However, there was no clear difference between controls and low exposure group. To confirm this tendency, a PCA model only for the control and low exposure groups was created, but no distinct boundary was observed between the two groups (Fig. 2B). A heatmap of 50 metabolites with the most contrasting patterns (ANOVA, p < 0.05) illustrates these findings (Fig. 2C). A clear, exposure-dependent metabolic dysregulation was observed: polar metabolites, including glutaric, gluconic and glutamic acids, as well as the bile acid LCA were upregulated in NOD mice exposed to a higher dose of POP mixture, while many SMs, PCs and LPCs were downregulated.

A and B PCA plots of metabolic profiles in NOD mice after exposure to a POP mixture for control (green), low exposure (blue) and high exposure (red) groups C Heatmap of 50 significantly-altered metabolites (ANOVA, p < 0.05) with the most contrasting patterns. Samples are sorted by the exposure group and metabolites clustered based on Ward’s clustering algorithm.
To investigate metabolites that were dysregulated already in the low exposure group, only in high exposure group and in both groups, a Venn diagram (Supplementary Figure A1) was plotted using jvenn plug-in (Bardou et al., 2014). Over one hundred metabolites (138) were significantly dysregulated (Tukey’s HSD, p < 0.05) in one or two exposure groups. Eighty-seven metabolites were commonly dysregulated in high compared to low exposure group and controls (Supplementary Table A3). Five metabolites common to all three comparison groups and five other metabolites common to both exposure groups were all altered. Thus, a Venn diagram confirmed that homeostasis was maintained to a much higher degree when NOD mice were exposed to a lower dose of POP mixture.
3.3 Interaction between BAs and other metabolites
Next, we studied the bile acid – metabolome interactions. While the majority of the BAs showed a negative correlation pattern with multiple lipid classes, the Spearman’s correlation indicated that a secondary bile acid, LCA, was positively associated with ceramides, diacylglycerol and saturated triglycerides, and negatively associated with phosphatidylcholines, lysophosphatidylcholines, sphingomyelins and polyunsaturated triglycerides (Fig. 3).

Spearman’s correlations between bile acids and lipid classes in NOD mouse blood serum samples. Significant correlations marked with *p < 0.05, **p < 0.01 and ***p < 0.001
Among the polar metabolites, aspartic, stearic and arachidonic acids had the strongest positive correlation with several secondary bile acids, such as deoxycholic acid (DCA) and its conjugates. Conversely, LCA showed inverse correlation to cholesterol, amino acids and free fatty acids, including serine, threonine, and aspartic, stearic and arachidonic acids, while 3-hydroxybutyric, fumaric, gluconic and glutamic acids were positively correlated with LCA (Supplementary Figure A2).
3.4 Pathway analysis
A pathway analysis, carried out in Metaboanalyst 5.0 using the Mus musculus Kyoto encyclopedia of genes and genomes (KEGG) pathway library and discrete classification, revealed 15 significantly-altered metabolic pathways (p < 0.05) (Table 3).
A graphic representation of altered pathways (Fig. 4) shows metabolic connections between the following downregulated classes of metabolites: (a) α-amino acids derived from oxaloacetate, (b) saturated and polyunsaturated long chain fatty acids, (c) phosphatidylcholines, (d) lysophosphatidylcholines, (e) sphingomyelins and (f) bile acids (except for increased LCA). At the same time, metabolites involved in the tricarboxylic acid cycle, glucose metabolism, and triglycerides were elevated.

Pathway analysis diagram representing, significantly-elevated metabolites/lipid classes in red (p < 0.05) and pink (0.05 < p < 0.1), as well as downregulated metabolite groups in blue (p < 0.05) and light blue (0.05 < p < 0.1), in mice after exposure to POPs Metabolites showing no significant changes are marked in light gray
4 Discussion
In the present study, we investigated profiles of polar metabolites, bile acids and lipids in female NOD mice that were pre- and postnatally exposed to a POP mixture containing PCBs, OCPs, BFRs and PFAS. Exposure to the highest dose tested of these organic pollutants caused more prominent alterations in metabolic pathways associated with bile acid, glucose, TCA, fatty acid, glycerophospholipid, sphingolipid and glycerolipid metabolism compared to the lower dose administered.
This study also had some limitations. The relatively low number of mice in control, low- and high-exposure groups (8, 7 and 7 respectively) may be seen as one of the limitations of this study. However, in animal studies this number of animals is usually considered sufficient, especially due to current recommendations of minimizing the number of animals. Another study limitation is that the metabolism of mice is not fully reflecting the metabolism in humans, therefore the results of this study cannot be extrapolated directly to human metabolism even if we observed similar alterations in humans (McGlinchey et al., 2020). Another limitation is that only female NOD mice were used in the exposure study. The female NOD mice develop spontaneous disease also without any exposure and thus, comparison of diabetic versus non-diabetic animals was not feasible in this study set-up.
The POP mixture used in the present study contained a mixture of POPs at concentration ratios mimicking the exposure profiles in humans (Berntsen et al., 2017). Although the POP concentrations used in this exposure study were higher than human estimated daily intake (EDI) values, the actual circulating PFAS concentrations in the serum of the NOD mice were in the range of human exposure levels. Even in the highest exposure group, PFAS concentrations were comparable with detected levels in occupational exposure studies (Zhou et al., 2014). It should be noted that in the control group not intentionally exposed to the POPs, low levels of PFAS could be detected, possibly due to presence of PFAS in the cage- and bedding materials, dust, or water dispenser.
The terminal half-lives for PFOS and PFOA differ between days for mice (Chang et al., 2012; Lou et al., 2009) and years for humans (Bartell et al., 2010; Brede et al., 2010; Li et al., 2020b; Olsen & Zobel, 2007). Due to these large differences in metabolic processes, including liver metabolism of xenobiotics between humans and rodents (Walton et al., 2001) and the rapid xenobiotic metabolism in mice compared to humans (Martignoni et al., 2006), it was necessary to apply high initial exposure levels. Yet, daily exposure doses of individual POPs for both low and high exposure NOD mouse groups were generally lower than previously established no adverse effect levels (NOAEL) for single compounds and total concentration of organochlorines in both exposure groups was below the levels that previously was shown to induce toxicity in standard toxicity tests (Bowers et al., 2004; Darnerud, 2003; DeWitt et al., 2019).
In our study, POP exposure in NOD mice was associated with decreased levels of several phospholipids, including particularly LPCs, PCs and SMs. Similar lipid changes have been reported in children who later developed islet autoimmunity (Johnson et al., 2019) and T1D (Oresic et al., 2008). In a recent study, downregulated SMs were associated with renal impairment and all-cause mortality in T1D (Tofte et al., 2019). In animal models, elevated LPC levels have also been observed in insulin autoantibody-positive female NOD mice that did not develop diabetes compared with progressors (Sysi-Aho et al., 2011). Interestingly, LPCs have been reported to lower blood glucose levels and activate adipocyte glucose uptake in T1D and T2D mouse models (Yea et al., 2009), also enhancing insulin secretion (Soga et al., 2005).
Another prominent lipid profile change observed in this study was elevated TG levels in the exposed mouse groups, especially in TGs containing saturated or monosaturated fatty acyls. These alterations are also consistent with previous studies reporting increased plasma triglyceride levels in patients with poor glycemic control of T1D (Dullaart, 1995). Hypertriglyceridemia in T1D develops due to increased levels of circulating free fatty acids, which in turn promote a production of very low-density lipoporotein (Nikkila & Kekki, 1973). The Environmental Determinants of Diabetes in the Young (TEDDY) study also showed a decreased abundance of unsaturated triglycerides prior to appearance of autoantibodies (Li et al., 2020c). In a NOD mice study, non-treated animals which became diabetic had significantly increased levels of 3-hydroxybutyric acid, triglycerides and cholesterol compared to a group treated with immunosuppressive rapamycin, a drug that prevented the onset of T1D (Baeder et al., 1992). This is in agreement with our findings, which showed elevated concentrations of TGs and 3-hydroxybutyric acid in the exposed mice compared to a control group, indicating that exposure led to even further raised TG levels, and hypothetically could promote an earlier onset of T1D. Early signs of accelerated T1D development in these mice have also been reported, as indicated by reduced numbers of tissue-resident macrophages in pancreatic islets and a trend of reduced macrophage phagocytosis (Berntsen et al., 2018). Thus, exposure to the POP mixture reducing LPCs and SMs and increasing TG blood levels may be linked with the accelerated development of T1D in NOD mice, and the present in vivo study supports causality of previously observed association studies in humans.
We also observed changes in bile acid metabolism, with the majority of BAs being downregulated by POP exposure. The downregulation of BAs is likely to be due to the PFAS, as it has been reported both in animal models and in vivo studies as a suppressor of de novo synthesis of BAs via suppression of cholesterol 7 alpha-hydroxylase (CYP7A1), which controls the first, rate-limiting step in the formation of BAs from cholesterol through the primary pathway (Beggs et al., 2016; Behr et al., 2020; Schlezinger et al., 2020). The only upregulated BA was LCA, a secondary bile acid. Bile acids have an important role in the regulation of lipid and glucose metabolism. Given this, we investigated whether the changes in the bile acid pool due to exposure were associated with changes in lipid metabolism. Indeed, we observed that LCA had a very strong positive correlation with those lipids that are classified as lipotoxic, namely, CEs, DGs and TGs containing saturated fatty acyls, while showing strong negative association with SMs and ether phosphatidylcholines. LCA has been previously associated with hepatotoxicity and decreased LPC and SM levels in mice (Matsubara et al., 2011), and also with the alteration of gene expression involved in cholesterol and phospholipid homeostasis, leading to an inflammation and liver injury (Dionne et al., 1994). A counterintuitive dysregulation pattern where 11 BAs were dysregulated in high exposure versus low exposure groups compared to only two dysregulated bile acids in high exposure vs control group may be explained by the observation that in the low exposure group, BAs showed upregulation trend, whereas in the high exposure group they were significantly downregulated (Supplementary Figure A3), suggesting a non-monotonic response pattern. Observed alteration of bile acids in our study may explain the dysregulation of specific lipid classes that were associated with T1D development in human studies (McGlinchey et al., 2020; Oresic et al., 2008).
Changes in the overall NOD mice bile acid pool were also observed. Both conjugated secondary (GDCA, GUDCA, TDCA, TDHCA, TLCA, TUDCA) and primary (GCA, GCDCA, TabMCA, TCA, TCDCA, TwMCA) bile acids were downregulated, with the decrease of conjugated secondary BAs being more pronounced (Supplementary Figure A3). The decrease of secondary BAs produced from the primary bile acids by colonic bacteria may suggest an alteration in the gut microbiota due to the dietary POP exposure via food pellets. Indeed, earlier findings have shown that POP exposure is associated with decreased gut microbiome diversity (Iszatt et al., 2019).
We also observed elevated glucose and glycolic acid in the exposed mice, while only two tricarboxylic acid cycle metabolites were significantly increased, which indicate changes in central carbon metabolism. Glucose has previously demonstrated a regulation of the farnesoid X receptor expression in liver and caused alterations in lipid and bile acid metabolism in patients with insulin resistance or diabetes (Duran-Sandoval et al., 2004). Previous studies have also associated prenatal POP exposure with adiposity and disturbances in glucose metabolism (Thomas et al., 2007). Particularly, PFOA has been found to be associated with higher levels of glycerol and 30-min glucose levels during an oral glucose tolerance test and PFAS with altered fatty acid and lipid metabolism (Chen et al., 2020). However, it should be noted that NOD mice develop insulin resistance even in the absence of high circulating glucose concentrations (Chaparro et al., 2006).
5 Conclusions
In conclusion, our mouse study showed that POP mixture exposure and, in particular measured blood PFAS levels, associated with alteration of multiple metabolic pathways. The metabolic changes observed were similar to those previously reported in human cohort studies investigating associations between exposure and biomarkers, including upregulation of glucose metabolites, triglycerides and lithocholic acid, and downregulation of long chain fatty acids, and several lipid classes including PCs, LPCs and SMs. Yet, the inconsistent findings in animal and epidemiological studies call for further investigations to better understand the associations between PFAS exposure and T1D development, including the underlying mechanisms. Combination of exposure studies in in vivo and in vitro models are needed to characterize the mechanisms, including investigation of metabolic changes not only in circulation but also in the liver and in feces. The latter is crucial as the exposure can have both direct impact of metabolism, but also an indirect impact via gut microbiota and gut microbial metabolites. In summary, our results suggest that observed metabolic alterations caused by exposure to the POP mixture can be used as potential biomarkers for T1D progression in exposure related studies.
References
Bach, J. F. (2001). Protective role of infections and vaccinations on autoimmune diseases. Journal of Autoimmunity, 16, 347–353.
Baeder, W. L., Sredy, J., Sehgal, S. N., Chang, J. Y., & Adams, L. M. (1992). Rapamycin prevents the onset of insulin-dependent diabetes mellitus (IDDM) in NOD mice. Clinical and Experimental Immunology, 89, 174–178.
Bao, M., Yang, Y., Jun, H. S., & Yoon, J. W. (2002). Molecular mechanisms for gender differences in susceptibility to T cell-mediated autoimmune diabetes in nonobese diabetic mice. The Journal of Immunology, 168, 5369–5375.
Bardou, P., Mariette, J., Escudie, F., Djemiel, C., & Klopp, C. (2014). jvenn: An interactive Venn diagram viewer. BMC Bioinformatics, 15, 293.
Bartell, S. M., Calafat, A. M., Lyu, C., Kato, K., Ryan, P. B., & Steenland, K. (2010). Rate of decline in serum PFOA concentrations after granular activated carbon filtration at two public water systems in Ohio and West Virginia. Environmental Health Perspectives, 118, 222–228.
Bassler, J., Ducatman, A., Elliott, M., Wen, S., Wahlang, B., Barnett, J., & Cave, M. C. (2019). Environmental perfluoroalkyl acid exposures are associated with liver disease characterized by apoptosis and altered serum adipocytokines. Environmental Pollution, 247, 1055–1063.
Beggs, K. M., McGreal, S. R., McCarthy, A., Gunewardena, S., Lampe, J. N., Lau, C., & Apte, U. (2016). The role of hepatocyte nuclear factor 4-alpha in perfluorooctanoic acid- and perfluorooctanesulfonic acid-induced hepatocellular dysfunction. Toxicology and Applied Pharmacology, 304, 18–29.
Behr, A. C., Kwiatkowski, A., Stahlman, M., Schmidt, F. F., Luckert, C., Braeuning, A., & Buhrke, T. (2020). Impairment of bile acid metabolism by perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in human HepaRG hepatoma cells. Archives of Toxicology, 94, 1673–1686.
Berntsen, H. F., Berg, V., Thomsen, C., Ropstad, E., & Zimmer, K. E. (2017). The design of an environmentally relevant mixture of persistent organic pollutants for use in in vivo and in vitro studies. Journal of Toxicology and Environmental Health, 80, 1002–1016.
Berntsen, H. F., Bolling, A. K., Bjorklund, C. G., Zimmer, K., Ropstad, E., Zienolddiny, S., Becher, R., Holme, J. A., Dirven, H., Nygaard, U. C., & Bodin, J. (2018). Decreased macrophage phagocytic function due to xenobiotic exposures in vitro, difference in sensitivity between various macrophage models. Food and Chemical Toxicology, 112, 86–96.
Bodin, J., Groeng, E. C., Andreassen, M., Dirven, H., & Nygaard, U. C. (2016). Exposure to perfluoroundecanoic acid (PFUnDA) accelerates insulitis development in a mouse model of type 1 diabetes. Toxicology Reports, 3, 664–672.
Bodin, J., Stene, L. C., & Nygaard, U. C. (2015). Can exposure to environmental chemicals increase the risk of diabetes type 1 development? Biomed Research International. https://doi.org/10.1155/2015/208947
Borg, D., Lund, B. O., Lindquist, N. G., & Hakansson, H. (2013). Cumulative health risk assessment of 17 perfluoroalkylated and polyfluoroalkylated substances (PFASs) in the Swedish population. Environment International, 59, 112–123.
Bowers, W. J., Nakai, J. S., Chu, I., Wade, M. G., Moir, D., Yagminas, A., Gill, S., Pulido, O., & Meuller, R. (2004). Early developmental neurotoxicity of a PCB/organochlorine mixture in rodents after gestational and lactational exposure. Toxicological Sciences, 77, 51–62.
Brede, E., Wilhelm, M., Goen, T., Muller, J., Rauchfuss, K., Kraft, M., & Holzer, J. (2010). Two-year follow-up biomonitoring pilot study of residents’ and controls’ PFC plasma levels after PFOA reduction in public water system in Arnsberg, Germany. International Journal of Hygiene and Environmental Health, 213, 217–223.
Cao, Y. G., Winneke, G., Wilhelm, M., Wittsiepe, J., Lemm, F., Furst, P., Ranft, U., Imohl, M., Kraftg, M., Oesch-Bartlomowicz, B., & Kramer, U. (2008). Environmental exposure to dioxins and polychlorinated biphenyls reduce levels of gonadal hormones in newborns: Results from the Duisburg cohort study. International Journal of Hygiene and Environmental Health, 211, 30–39.
Casey, A. C., Berger, D. F., Lombardo, J. P., Hunt, A., & Quimby, F. (1999). Aroclor 1242 inhalation and ingestion by Sprague-Dawley rats. Journal of Toxicology and Environmental Health-Part a-Current Issues, 56, 311–342.
Castillo, S., Mattila, I., Miettinen, J., Oresic, M., & Hyotylainen, T. (2011). Data analysis tool for comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry. Analytical Chemistry, 83, 3058–3067.
Chang, S. C., Noker, P. E., Gorman, G. S., Gibson, S. J., Hart, J. A., Ehresman, D. J., & Butenhoff, J. L. (2012). Comparative pharmacokinetics of perfluorooctanesulfonate (PFOS) in rats, mice, and monkeys. Reproductive Toxicology, 33, 428–440.
Chaparro, R. J., Konigshofer, Y., Beilhack, G. F., Shizuru, J. A., McDevitt, H. O., & Chien, Y. H. (2006). Nonobese diabetic mice express aspects of both type 1 and type 2 diabetes. Proceedings of the National Academy of Sciences of the United States of America, 103, 12475–12480.
Chatenoud, L., Warncke, K., & Ziegler, A. G. (2012). Clinical Immunologic Interventions for the Treatment of Type 1 Diabetes. Cold Spring Harbor Perspectives in Medicine, 2, a007716.
Chen, J. W., Wang, S. L., Liao, P. C., Chen, H. Y., Ko, Y. C., & Lee, C. C. (2008). Relationship between insulin sensitivity and exposure to dioxins and polychlorinated biphenyls in pregnant women. Environmental Research, 107, 245–253.
Chen, Z. H., Yang, T. Y., Walker, D. I., Thomas, D. C., Qiu, C. Y., Chatzi, L., Alderete, T. L., Kim, J. S., Conti, D. V., Breton, C. V., Liang, D. H., Hauser, E. R., Jones, D. P., & Gilliland, F. D. (2020). Dysregulated lipid and fatty acid metabolism link perfluoroalkyl substances exposure and impaired glucose metabolism in young adults. Environment International, 145, 106091.
Conway, B., Innes, K. E., & Long, D. (2016). Perfluoroalkyl substances and beta cell deficient diabetes. Journal of Diabetes and Its Complications, 30, 993–998.
Darnerud, P. O. (2003). Toxic effects of brominated flame retardants in man and in wildlife. Environment International, 29, 841–853.
DeWitt, J. C., Blossom, S. J., & Schaider, L. A. (2019). Exposure to per-fluoroalkyl and polyfluoroalkyl substances leads to immunotoxicity: Epidemiological and toxicological evidence. Journal of Exposure Science and Environmental Epidemiology, 29, 148–156.
Dionne, S., Tuchweber, B., Plaa, G. L., & Yousef, I. M. (1994). Phase-I and Phase-II metabolism of lithocholic acid in hepatic acinar zone-3 necrosis : Evaluation in rats by combined radiochromatography and gas-liquid-chromatography mass-spectrometry. Biochemical Pharmacology, 48, 1187–1197.
Dullaart, R. P. F. (1995). Plasma-lipoprotein abnormalities in type-1 (insulin-dependent) Diabetes-Mellitus. Netherlands Journal of Medicine, 46, 44–54.
Duran-Sandoval, D., Mautino, G., Martin, G. V., Percevault, F., Barbier, O., Fruchart, J. C., Kuipers, F., & Staels, B. (2004). Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes, 53, 890–898.
Gascon, M., Vrijheid, M., Martinez, D., Forns, J., Grimalt, J. O., Torrent, M., & Sunyer, J. (2011). Effects of pre and postnatal exposure to low levels of polybromodiphenyl ethers on neurodevelopment and thyroid hormone levels at 4 years of age. Environment International, 37, 605–611.
Group TS. (2008). The environmental determinants of diabetes in the young (TEDDY) Study. Annals of the New York Academy of Sciences, 1150, 1–13.
Harjutsalo, V., Sund, R., Knip, M., & Groop, P. H. (2013). Incidence of type 1 diabetes in Finland. Jama-Journal of the American Medical Association, 310, 427–428.
Harrad, S., Wijesekera, R., Hunter, S., Halliwell, C., & Baker, R. (2004). Preliminary assessment of UK human dietary and inhalation exposure to polybrominated diphenyl ethers. Environmental Science and Technology, 38, 2345–2350.
Haug, L. S., Huber, S., Becher, G., & Thomsen, C. (2011). Characterisation of human exposure pathways to perfluorinated compounds - Comparing exposure estimates with biomarkers of exposure. Environment International, 37, 687–693.
Hazrati, S., & Harrad, S. (2006). Causes of variability in concentrations of polychlorinated biphenyls and polybrominated diphenyl ethers in indoor air. Environmental Science and Technology, 40, 7584–7589.
HBM4EU. (2020). Dashboard European Human Biomonitoring Data for visualisation of aggregated data., Flemish Institute for Technological Research (VITO), Mol, Belgium.
Heilmann, C., Grandjean, P., Weihe, P., Nielsen, F., & Budtz-Jorgensen, E. (2006). Reduced antibody responses to vaccinations in children exposed to polychlorinated biphenyls. Plos Medicine, 3, 1352–1359.
Hites, R. A., Foran, J. A., Carpenter, D. O., Hamilton, M. C., Knuth, B. A., & Schwager, S. J. (2004). Global assessment of organic contaminants in farmed salmon. Science, 303, 226–229.
Hwang, H. M., Park, E. K., Young, T. M., & Hammock, B. D. (2008). Occurrence of endocrine-disrupting chemicals in indoor dust. Science of the Total Environment, 404, 26–35.
Iszatt, N., Janssen, S., Lenters, V., Dahl, C., Stigum, H., Knight, R., Mandal, S., Peddada, S., Gonzalez, A., Midtvedt, T., & Eggesbo, M. (2019). Environmental toxicants in breast milk of Norwegian mothers and gut bacteria composition and metabolites in their infants at 1 month. Microbiome. https://doi.org/10.1186/s40168-019-0645-2
Johnson, R. K., Vanderlinden, L., DeFelice, B. C., Kechris, K., Uusitalo, U., Fiehn, O., Sontag, M., Crume, T., Beyerlein, A., Lernmark, A., Toppari, J., Ziegler, A. G., She, J. X., Hagopian, W., Rewers, M., Akolkar, B., Krischer, J., Virtanen, S. M., & Norris, J. M. (2019). Metabolite-related dietary patterns and the development of islet autoimmunity. Scientific Reports. https://doi.org/10.1038/s41598-019-51251-4
JRC. (2018). Something from nothing? Ensuring the safety of chemical mixtures. JRC Science for policy brief. Based on JRC110683.
Karrman, A., Ericson, I., van Bavel, B., Darnerud, P. O., Aune, M., Glynn, A., Lignell, S., & Lindstrom, G. (2007). Exposure of perfluorinated chemicals through lactation: Levels of matched human milk and serum and a temporal trend, 1996–2004, in Sweden. Environmental Health Perspectives, 115, 226–230.
Karrman, A., van Bavel, B., Jarnberg, U., Hardell, L., & Lindstrom, G. (2006). Perfluorinated chemicals in relation to other persistent organic pollutants in human blood. Chemosphere, 64, 1582–1591.
Kelemen, K. (2004). The role of T cells in beta cell damage in NOD mice and humans. Immunology of Type 1 Diabetes Second Edition, 552, 117–128.
Lau, C. (2012). Molecular, clinical and environmental toxicology. Volume 3: Environmental toxicology. Springer.
Lee, D. H., Lee, I. K., Jin, S. H., Steffes, M., & Jacobs, D. R. (2007). Association between serum concentrations of persistent organic pollutants and insulin resistance among nondiabetic adults. Diabetes Care, 30, 622–628.
Lee, D. H., Steffes, M. W., Sjodin, A., Jones, R. S., Needham, L. L., & Jacobs, D. R. (2010). Low dose of some persistent organic pollutants predicts type 2 diabetes: A nested case-control study. Environmental Health Perspectives, 118, 1235–1242.
Li, J., Cai, D., Chu, C., Li, Q. Q., Zhou, Y., Hu, L. W., Yang, B. Y., Dong, G. H., Zeng, X. W., & Chen, D. (2020a). Transplacental transfer of per- and polyfluoroalkyl substances (PFASs): Differences between preterm and full-term deliveries and associations with placental transporter mRNA expression. Environmental Science and Technology, 54, 5062–5070.
Li, Q., Parikh, H., Butterworth, M. D., Lernmark, A., Hagopian, W., Rewers, M., She, J. X., Toppari, J., Ziegler, A. G., Akolkar, B., Fiehn, O., Fan, S. L., Krischer, J. P., & Grp, T. S. (2020b). Longitudinal metabolome-wide signals prior to the appearance of a first islet autoantibody in children participating in the TEDDY study. Diabetes, 69, 465–476.
Li, Y., Barregard, L., Xu, Y. Y., Scott, K., Pineda, D., Lindh, C. H., Jakobsson, K., & Fletcher, T. (2020c). Associations between perfluoroalkyl substances and serum lipids in a Swedish adult population with contaminated drinking water. Environmental Health. https://doi.org/10.1186/s12940-020-00588-9
Lin, C. Y., Chen, P. C., Lin, Y. C., & Lin, L. Y. (2009). Association among serum perfluoroalkyl chemicals, glucose homeostasis, and metabolic syndrome in adolescents and adults. Diabetes Care, 32, 702–707.
Lopez-Espinosa, M. J., Mondal, D., Armstrong, B., Bloom, M. S., & Fletcher, T. (2012). Thyroid function and perfluoroalkyl acids in children living near a chemical plant. Environmental Health Perspectives, 120, 1036–1041.
Lou, I. C., Wambaugh, J. F., Lau, C., Hanson, R. G., Lindstrom, A. B., Strynar, M. J., Zehr, R. D., Setzer, R. W., & Barton, H. A. (2009). Modeling single and repeated dose pharmacokinetics of PFOA in mice. Toxicological Sciences, 107, 331–341.
Mamsen, L. S., Bjorvang, R. D., Mucs, D., Vinnars, M. T., Papadogiannakis, N., Lindh, C. H., Andersen, C. Y., & Damdimopoulou, P. (2019). Concentrations of perfluoroalkyl substances (PFASs) in human embryonic and fetal organs from first, second, and third trimester pregnancies. Environment International, 124, 482–492.
Mandalakis, M., Stephanou, E. G., Horii, Y., & Kannan, K. (2008). Emerging contaminants in car interiors: Evaluating the impact of airborne PBDEs and PBDD/Fs. Environmental Science and Technology, 42, 6431–6436.
Martignoni, M., Groothuis, G. M. M., & de Kanter, R. (2006). Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opinion on Drug Metabolism & Toxicology, 2, 875–894.
Matsubara, T., Tanaka, N., Patterson, A. D., Cho, J. Y., Krausz, K. W., & Gonzalez, F. J. (2011). Lithocholic acid disrupts phospholipid and sphingolipid homeostasis leading to cholestasis in mice. Hepatology, 53, 1282–1293.
McGlinchey, A., Sinioja, T., Lamichhane, S., Sen, P., Bodin, J., Siljander, H., Dickens, A. M., Geng, D. W., Carlsson, C., Duberg, D., Ilonen, J., Virtanen, S. M., Dirven, H., Berntsen, H. F., Zimmer, K., Nygaard, U. C., Oresic, M., Knip, M., & Hyotylainen, T. (2020). Prenatal exposure to perfluoroalkyl substances modulates neonatal serum phospholipids, increasing risk of type 1 diabetes. Environment International. https://doi.org/10.1016/j.envint.2020.105935
Mol, N. M., Sorensen, N., Weihe, P., Andersson, A. M., Jorgensen, N., Skakkebaek, N. E., Keiding, N., & Grandjean, P. (2002). Spermaturia and serum hormone concentrations at the age of puberty in boys prenatally exposed to polychlorinated biphenyls. European Journal of Endocrinology, 146, 357–363.
Mullerova, D., & Kopecky, J. (2007). White adipose tissue: Storage and effector site for environmental pollutants. Physiological Research, 56, 375–381.
Nikkila, E. A., & Kekki, M. (1973). Plasma triglyceride transport kinetics in diabetes mellitus. Metabolism, 22, 1–22.
Nygren, H., Seppanen-Laakso, T., Castillo, S., Hyotylainen, T., & Oresic, M. (2011). Liquid chromatography-mass spectrometry (LC-MS)-based lipidomics for studies of body fluids and tissues. Methods in Molecular Biology, 708, 247–257.
Olsen, G. W., & Zobel, L. R. (2007). Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. International Archives of Occupational and Environmental Health, 81, 231–246.
Oresic, M., Simell, S., Sysi-Aho, M., Nanto-Salonen, K., Seppanen-Laakso, T., Parikka, V., Katajamaa, M., Hekkala, A., Mattila, I., Keskinen, P., Yetukuri, L., Reinikainen, A., Lahde, J., Suortti, T., Hakalax, J., Simell, T., Hyoty, H., Veijola, R., Ilonen, J., … Simell, O. (2008). Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. Journal of Experimental Medicine, 205, 2975–2984.
Park, H. Y., Hertz-Picciotto, I., Petrik, J., Palkovicova, L., Kocan, A., & Trnovec, T. (2008). Prenatal PCB exposure and thymus size at birth in neonates in eastern Slovakia. Environmental Health Perspectives, 116, 104–109.
Patterson, C. C., Karuranga, S., Salpea, P., Saeedi, P., Dahlquist, G., Soltesz, G., & Ogle, G. D. (2019). Worldwide estimates of incidence, prevalence and mortality of type 1 diabetes in children and adolescents: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Research and Clinical Practice, 157, 107842.
Pluskal, T., Castillo, S., Villar-Briones, A., & Oresic, M. (2010). MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. https://doi.org/10.1186/1471-2105-11-395
Porpora, M. G., Lucchini, R., Abballe, A., Ingelido, A. M., Valentini, S., Fuggetta, E., Cardi, V., Ticino, A., Marra, V., Fulgenzi, A. R., & De Felip, E. (2013). Placental transfer of persistent organic pollutants: A preliminary study on mother-newborn pairs. International Journal of Environmental Research and Public Health, 10, 699–711.
Predieri, B., Iughetti, L., Guerranti, C., Bruzzi, P., Perra, G., & Focardi, S. E. (2015). High levels of perfluorooctane sulfonate in children at the onset of diabetes. International Journal of Endocrinology. https://doi.org/10.1155/2015/234358
Regnell, S. E., & Lernmark, A. (2013). The environment and the origins of islet autoimmunity and Type 1 diabetes. Diabetic Medicine, 30, 155–160.
Rook, G. A. W. (2012). Hygiene hypothesis and autoimmune diseases. Clinical Reviews in Allergy and Immunology, 42, 5–15.
Salihovic, S., Dickens, A. M., Schoultz, I., Fart, F., Sinisalu, L., Lindeman, T., Halfvarson, J., Oresic, M., & Hyotylainen, T. (2020). Simultaneous determination of perfluoroalkyl substances and bile acids in human serum using ultra-high-performance liquid chromatography-tandem mass spectrometry. Analytical and Bioanalytical Chemistry, 412, 2251–2259.
Salo, H. M., Koponen, J., Kiviranta, H., Rantakokko, P., Honkanen, J., Harkonen, T., Ilonen, J., Virtanen, S. M., Tillmann, V., Knip, M., & Vaarala, O. (2019). No evidence of the role of early chemical exposure in the development of -cell autoimmunity. Environmental Science and Pollution Research, 26, 1370–1378.
Schlezinger, J. J., Puckett, H., Oliver, J., Nielsen, G., Heiger-Bernays, W., & Webster, T. F. (2020). Perfluorooctanoic acid activates multiple nuclear receptor pathways and skews expression of genes regulating cholesterol homeostasis in liver of humanized PPAR alpha mice fed an American diet. Toxicology and Applied Pharmacology. https://doi.org/10.1101/2020.01.30.926642
Sinisalu, L., Sen, P., Salihovic, S., Virtanen, S. M., Hyoty, H., Ilonen, J., Toppari, J., Veijola, R., Oresic, M., Knip, M., & Hyotylainen, T. (2020). Early-life exposure to perfluorinated alkyl substances modulates lipid metabolism in progression to celiac disease. Environmental Research, 188, 109864.
Soga, T., Ohishi, T., Matsui, T., Saito, T., Matsumoto, M., Takasaki, J., Matsumoto, S., Kamohara, M., Hiyama, H., Yoshida, S., Momose, K., Ueda, Y., Matsushime, H., Kobori, M., & Furuichi, K. (2005). Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochemical and Biophysical Research Communications, 326, 744–751.
Stapleton, H. M., Eagle, S., Anthopolos, R., Wolkin, A., & Miranda, M. L. (2011). Associations between polybrominated diphenyl ether (PBDE) flame retardants, phenolic metabolites, and thyroid hormones during pregnancy. Environmental Health Perspectives, 119, 1454–1459.
Stene, L. C., & Gale, E. A. M. (2013). The prenatal environment and type 1 diabetes. Diabetologia, 56, 1888–1897.
Sysi-Aho, M., Ermolov, A., Gopalacharyulu, P. V., Tripathi, A., Seppanen-Laakso, T., Maukonen, J., Mattila, I., Ruohonen, S. T., Vahatalo, L., Yetukuri, L., Harkonen, T., Lindfors, E., Nikkila, J., Ilonen, J., Simell, O., Saarela, M., Knip, M., Kaski, S., Savontaus, E., & Oresic, M. (2011). Metabolic regulation in progression to autoimmune diabetes. Plos Computational Biology, 7, e1002257.
Thomas, C., Hypponen, E., & Power, C. (2007). Prenatal exposures and glucose metabolism in adulthood - Are effects mediated through birth weight and adiposity? Diabetes Care, 30, 918–924.
Tofte, N., Suvitaival, T., Ahonen, L., Winther, S. A., Theilade, S., Frimodt-Moller, M., Ahluwalia, T. S., & Rossing, P. (2019). Lipidomic analysis reveals sphingomyelin and phosphatidylcholine species associated with renal impairment and all-cause mortality in type 1 diabetes. Scientific Reports. https://doi.org/10.1038/s41598-019-52916-w
Vasiliu, O., Cameron, L., Gardiner, J., DeGuire, P., & Karmaus, W. (2006). Polybrominated biphenyls, polychlorinated biphenyls, body weight, and incidence of adult-onset diabetes mellitus. Epidemiology, 17, 352–359.
Walton, K., Dorne, J. L., & Renwick, A. G. (2001). Uncertainty factors for chemical risk assessment: Interspecies differences in the in vivo pharmacokinetics and metabolism of human CYP1A2 substrates. Food and Chemical Toxicology, 39, 667–680.
Wilford, B. H., Harner, T., Zhu, J. P., Shoeib, M., & Jones, K. C. (2004). Passive sampling survey of polybrominated diphenyl ether flame retardants in indoor and outdoor air in Ottawa, Canada: Implications for sources and exposure. Environmental Science and Technology, 38, 5312–5318.
Xia, J. G., & Wishart, D. S. (2011). Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nature Protocols, 6, 743–760.
Yea, K., Kim, J., Yoon, J. H., Kwon, T., Kim, J. H., Lee, B. D., Lee, H. J., Lee, S. J., Kim, J. I., Lee, T. G., Baek, M. C., Park, H. S., Park, K. S., Ohba, M., Suh, P. G., & Ryu, S. H. (2009). Lysophosphatidylcholine activates adipocyte glucose uptake and lowers blood glucose levels in murine models of diabetes. Journal of Biological Chemistry, 284, 33833–33840.
Zhou, Z., Shi, Y. L., Vestergren, R., Wang, T., Liang, Y., & Cai, Y. Q. (2014). Highly elevated serum concentrations of perfluoroalkyl substances in fishery employees from Tangxun Lake, China. Environmental Science and Technology, 48, 3864–3874.
Zota, A. R., Park, J. S., Wang, Y., Petreas, M., Zoeller, R. T., & Woodruff, T. J. (2011). Polybrominated diphenyl ethers, hydroxylated polybrominated diphenyl ethers, and measures of thyroid function in second trimester pregnant women in California. Environmental Science and Technology, 45, 7896–7905.
Acknowledgements
We would like to acknowledge Dr. Aidan McGlinchey for proofreading the article and helpful scientific discussions. This study was supported by funding from Vetenskapsrådet (to T.H. and M.O.; grants no. 2016-05176 and 2020-03674), Formas (to T.H. and M.O.; grant no. 2019-00869) and Academy of Finland (grant no. 333981 to M.O.), as well as by funding from the Norwegian Research Council, project number 213060 and the Norwegian Institute of Public Health. The metabolomics datasets in this study were submitted to MetaboLights, under accession number MTBLS875.
Funding
Open access funding provided by Örebro University.
Author information
Authors and Affiliations
Contributions
T.H. and M.O. proposed the metabolomics studies in NOD mice and they supervised data analysis and integration, and the interpretation of analytical outcomes. T.S. and D.D. performed the laboratory analyses and T.S. analyzed the data. T.H. supervised metabolomic and PFAS analyses. J.B., H.D. and U.C.N. conducted the studies in NOD mice, while H.F.B. and K.Z. assembled the internal relationship of chemicals in the feed for POP-exposure. T.S. wrote the first version of the paper under the supervision of M.O. and T.H. All authors approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sinioja, T., Bodin, J., Duberg, D. et al. Exposure to persistent organic pollutants alters the serum metabolome in non-obese diabetic mice. Metabolomics 18, 87 (2022). https://doi.org/10.1007/s11306-022-01945-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-022-01945-0