
Abstract
Lung cancer causes more deaths in men and women than any other cancer related disease. Currently, few effective strategies exist to predict how patients will respond to treatment. We evaluated the serum metabolomic profiles of 25 lung cancer patients undergoing chemotherapy ± radiation to evaluate the feasibility of metabolites as temporal biomarkers of clinical outcomes. Serial serum specimens collected prospectively from lung cancer patients were analyzed using both nuclear magnetic resonance (1H-NMR) spectroscopy and gas chromatography mass spectrometry (GC–MS). Multivariate statistical analysis consisted of unsupervised principal component analysis or orthogonal partial least squares discriminant analysis with significance assessed using a cross-validated ANOVA. The metabolite profiles were reflective of the temporal distinction between patient samples before during and after receiving therapy (1H-NMR, p < 0.001: and GC–MS p < 0.01). Disease progression and survival were strongly correlative with the GC–MS metabolite data whereas stage and cancer type were associated with 1H-NMR data. Metabolites such as hydroxylamine, tridecan-1-ol, octadecan-1-ol, were indicative of survival (GC–MS p < 0.05) and metabolites such as tagatose, hydroxylamine, glucopyranose, and threonine that were reflective of progression (GC–MS p < 0.05). Metabolite profiles have the potential to act as prognostic markers of clinical outcomes for lung cancer patients. Serial 1H-NMR measurements appear to detect metabolites diagnostic of tumor pathology, while GC–MS provided data better related to prognostic clinical outcomes, possibility due to physiochemical bias related to specific biochemical pathways. These results warrant further study in a larger cohort and with various treatment options.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Lung cancer is the leading cause of cancer related deaths in both men and women in the world with 1.8 million patients in 2012 and an estimated 1.6 million deaths. The majority of lung cancers are diagnosed at an advanced stage at which time all treatment is of palliative intent. Currently, there are no prognostic or predictive biomarkers to help ascertain which patients are most likely to benefit from chemotherapy or radiation (Sawyers 2008).
Metabolic profiling has been reported in small series in several types of cancers including lung (Fan et al. 2009; Carrola et al. 2011; Yang et al. 2010; Jordan et al. 2010), breast (Chen et al. 2009), ovarian (Odunsi et al. 2005), prostate (Sreekumar et al. 2009), pancreatic (Bathe et al. 2011) and colorectal (Farshidfar et al. 2012). In studies of lung cancer, most have explored the application of metabolomics for diagnosis comparing samples from lung cancer patients versus healthy volunteers (Fan et al. 2009; Carrola et al. 2011; Yang et al. 2010) or samples from lung cancer tissues versus surrounding non-cancerous tissues (Hori et al. 2011). These studies have demonstrated that many primary metabolites are found at higher levels in lung cancer tissues/patients versus controls. Additionally, others have examined and demonstrated that differences in the levels of metabolites can distinguish between tumour subtype (Jordan et al. 2010; Hori et al. 2011) and stage (Hori et al. 2011). Wedge et al. (2011) evaluated plasma from patients with small cell lung cancer and observed that particular metabolites were associated with worse survival outcomes. When 1H-NMR was used to evaluate metabolic changes in 3LL lung cancer tumours grown subcutaneously in mice who were treated with nitrosourea, alterations in metabolites were observed in the growth inhibition and growth recovery phase (Morvan and Demidem 2007). More recently, Deja et al. (2014) using 1H-NMR reported differences between COPD versus different stages of lung cancer, also differences between early and late stage NSCLC. Wen et al. (2013) utilized both GC–MS and LC–MS to reveal metabolic differences between patients with early stage adenocarcinoma of the lung and healthy controls. Similarly, Lokhav et al. (2013) looked at discriminating metabolomic features between patients with any stage of lung cancer versus healthy controls.
The majority of previous work concerns elucidation of diagnostic metabolite patterns or markers, however there have been only a few studies exploring the use of metabolomics technologies as prognostic markers. Here we examine metabolite profiles of lung cancer patients serially during the course of treatment utilizing comprehensive profiling by 1H-NMR and GC–MS. In this pilot study we establish the feasibility of characterizing the serum metabolic profiles of patients prior to, during, and in follow-up after receiving standard chemotherapy and/or radiation. We hypothesize that system-level analysis of serum metabolites may provide a potentially non-invasive approach to distinguish differences in physiologic state that may correlate with clinical outcomes.
2 Materials and methods
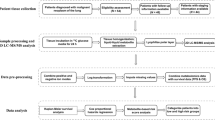
2.1 Subjects and sample collection
Any patient with a histologic or cytologically confirmed diagnosis of non-metastatic lung cancer undergoing standard chemotherapy ± radiation was eligible. Patients with synchronous malignancies or prior chemotherapy or radiation for any reason were excluded. Serum specimens were collected at pre-specified time-points: prior to treatment, during treatment, and at 6 months post-treatment. Blood was collected by venipuncture, centrifuged at 1500×g for 10 min at 4 °C and separated within 24 h of collection then stored at −80 °C until 1H-NMR and GC–MS analysis. Patients were not fasting at the time of blood sampling. In total, analyzed 134 serum samples were analyzed.
2.2 1H-NMR and GC–MS measurements
2.2.1 NMR sample preparation and data acquisition
1H-NMR spectroscopy was performed using a protocol previously described (Weljie et al. 2007). For NMR analysis, serum samples were thawed in ice. 350 μl of serum sample were filtered through a prewashed Nanasep 3K Omega Filter Eppendorf to remove high molecular weight (>3 kDa) compounds (e.g. large proteins, lipid complexes etc.). The filtrate was then centrifuged and buffered to a pH of 7.0 for analysis. Regular one-dimensional proton NMR spectra were obtained using a 600-MHz Bruker Ultrashield NMR spectrometer (Bruker Biospin, Milton, Canada). The spectra were acquired using a standard pulse sequence that had good water suppression characteristics and is commonly used for metabolite profiling of serum samples. Relaxation delay of 1 s was used; t1 was set to 4 μs and tm had a value of 100 ms. Initial samples for each batch were shimmed to ensure half-height line width of <1.1 Hz for the dimethyly-silapentane-sulphonate peak, calibrated to 0.0 ppm. Spectra were acquired with 1024 scans, then zero filled and Fourier transformed to 128 k data points using the Chenomx NMRSuite processor (Weljie et al. 2006). Additional 2-dimensional NMR experiments were performed for the purpose of confirming chemical shift assignments, including homonuclear total correlation spectroscopy (2D 1H-1H TOCSY) and heteronuclear single quantum coherence spectroscopy (2D 1H-13C HSQC), using standard Bruker pulse programs.
2.2.2 GC–MS sample preparation and data acquisition
Sample preparation for the GC–MS consists of three basic steps of deproteinization, methoxiamination and finally derivatization. Deproteinization is able to remove the interference from proteins such as albumin, which are present in a large concentration in blood. For GC–MS metabolite extraction, samples are thawed on ice and 50 μl of sample used for extraction using chloroform: methanol procedure. Aqueous metabolites will be dried under vacuum, and then derivatized using methoxyamine and MSTFA. Mass spectral data was acquired using a Water GCT premier GC-TOF–MS with an Agilent GC using a 30 m DB-5 column as previously described (Farshidfar et al. 2012).
2.2.3 1H-NMR and GC–MS data processing and statistical analysis
Raw data from 1H-NMR was processed and profiled using Chenomx NMR Suite 7.1 to a library of 55 compounds. 1H-NMR spectral data was evaluated using the strategy of ‘targeted profiling’ (Weljie et al. 2006). This allows quantification of metabolite concentrations in the samples. Raw data from GC–MS was imported to Metabolite Detector for peak detection (Hiller et al. 2009).
The pre-processed data was exported to multi-variate statistical analytical software SIMCA-P 13+ and analyzed with principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA). Univariate scaling is applied to overcome the dominating effect of higher intensity metabolite features. Thus scaling gives equal importance to all the components. Logarithmic transformation was also done before analysis to reduce the effect of data skewness.
PCA was performed to check the unsupervised segregation of the metabolome. OPLS-DA allowed us to discriminate between the different variables concerning the cancers in context. Model significance was assessed using a cross-validated ANOVA with p ≤ 0.05 considered significant. Variables were selected according to the VIP (variable influence on projection) as previously described (Weljie et al. 2007), which are reflective of the correlation of the metabolites towards different response. VIP > 1 was considered significant.
To elucidate the relative changes of significant metabolites before, during and after therapy, significance of analysis of microarrays (SAM) with time course analysis was performed (Zhang 2007). This analysis was applied independently to both GC–MS and 1H-NMR metabolite datasets (Denery et al. 2011). Metabolites were considered significant with a corrected p value <0.05 and false discovery rate of 10 % to account for multiple testing.
We further investigated if specific clinical and outcome factors, included variables such as cancer type (NSCLC and SCLC), cancer staging (stages 1–4), time-point(s) of sample collection, survival and progression could be predicted by the GC–MS and 1H-NMR datasets.
3 Results
3.1 Patient characteristics
Patient clinical and demographic characteristics of the patients are listed in Table 1. 18 (72 %) had non-small cell lung cancer, while 7 (18 %) had small cell lung cancer. The majority of patients had stage III disease and underwent treatment with concurrent chemoradiotherapy. The 2-year overall survival for the group was 53 %.
3.2 Metabolomics analysis
3.2.1 Temporal metabolomic analysis distinguish sera across treatment
Serum samples from each patient collected pre-treatment, during therapy, and post-treatment were subject to both NMR and GC–MS analysis. A total of 56 features were quantitatively assessed by NMR, and 106 features identified by GC–MS. Figure 1a is a box and whisker plot of the multivariate scores of OPLS-DA analysis of GC–MS metabolomic data from the three time-points. In this analysis, the discriminant analysis was naïve to the temporal nature of the data, however a defined temporal decrease in the scores from the first predicative component is apparent. Analysis of the OPLS-DA loadings reveals that the differences in timepoints was based on the differential abundance of glucopyranose, citric acid, butanoic acid, erythritol and ribitol between the three groups (N = 61, CV-ANOVA p = 0.0046, R2 = 0.186, Q2 = 0.128) (Table 2a.VIP > 1). Figure 1b presents a similar analysis of 1H-NMR metabolomic data. The differences were based on the changing level of metabolites 2-aminobutyrate, 2-oxoglutarate, threonine, methionine, creatinine and citrate between the three groups (N = 64 CV-ANOVA p < 0.001, R2 = 0.223, Q2 = 0.143) (Table 2b.VIP > 1). Interestingly metabolites such as threonine and citrate were part of up the 1H-NMR metabolite data; were not picked by the GCMS.

Metabolite bioprofiling facilitates discrimination between three groups of patient sample collected at pre-treatment, mid-therapy and post-treatment time points. Box and whisker plot reflective of three distinct time points based on scores of OPLS-DA model of a GC–MS and b NMR analysis of serum samples respectively; Heatmap showing clustering of metabolites based on time for both c NMR and GC–MS data respectively
In order to further probe the temporal nature of the data, we employed an analysis specific for time course data (SAM), which utilized the repeated measurements within individual patient samples. From the GCMS data, SAM analysis identified 8 differentially abundant metabolites across all treatment points (Fig. 1c). Of these named metabolites included 2-hydroxybutanoic acid, glucopyranose, citric acid, erythritol and ribitol. 1H-NMR metabolite data was able to identify three significant metabolites across the designated time points of sample collection. These included taurine, threonine and creatinine (Fig. 1c).
3.3 Relationship of metabolite data to clinical outcomes
3.3.1 GCMS metabolomic profiles facilitating prognostic evaluation of survival and progression from pre-treatment samples
From our preliminary O2-PLS models including all clinical covariates, we established that progression and survival were the two variables reflected by the GC–MS data (Fig. S1A). We then constructed individual OPLS-DA models for these covariates (Survival, Fig. S2B; Progression, Fig. S2C) based on the metabolic samples of pre-treatment samples only, i.e. naïve to any chemotherapy or radiotherapy. The survival model distinguishes the metabolite profile of patients who had died during the course of treatment from those who had survived treatment (N = 25, CV-ANOVA p = 0.0335, R2 = 0.388, Q2 = 0.285). This distinction was based on relative differences in metabolites such as tridecan-1-ol, octadecan-1-ol and hydroxylamine in that were abundant in patients who did not survive. Figure 2a is a box and whisker plots that show the distribution of scores as a function of deceased vs. survived therapy. A heatmap was constructed using hierarchical clustering (Fig. 2b). This data illustrates of metabolites such as glutamine, proline, valine, threonine and tyramine being differentially abundant in the population of patients who survived therapy where metabolites such as hydroxylamine and octadecan-1-ol being at a higher concentration in patients who did not survive therapy.

GC-MS metabolite bioprofiling facilitates prognostic evaluation of clinical outcomes based on survival and disease progression. a Box and whisker plot based on scores from OPLS-DA model of patient survival at pretreatment as a function of the eventual survival status: b Heat map showing clustering of metabolites with respect to patient survival (c, d) Progression: c as in a, with samples stratified by evidence of progression; d Heatmap showing progression-related metabolites; e Shared and unique structure (SUS) Plot, highlighting the strong relation between the two variables of disease progression and survival. The metabolites that line up along the diagonal running from the lower left corner to the upper right corner are common to both the patient progression and survival model
Similarly, a significant OPLS-DA model was built demonstrating a relationship between the GCMS metabolic profile and disease progression (Fig. 2c; Fig. S2C; N = 25, CV-ANOVA p < 0.05, R2 = 0.397, Q2 = 0.333). This difference was based on metabolites such as tagatose, hydroxylamine, glucopyranose, and threonine. A heat map illustrating the features involved in disease progression using SAM analysis to distinguish groups (Fig. 2d) demonstrates metabolites such as hydroxylamine are at relatively higher concentration in the group of patients who were noted to progress with the disease despite therapy, however glucopyranose and threonic acid were the metabolites was found to be at a relatively higher level in patients in whom the disease did not show signs of progression.
Clinically, progression and survival are related events, and thus in order to further understand the relationship between metabolite reflective of survival and disease progression a shared and unique structure (SUS) plot was constructed (Fig. 2e). The metabolites that line up along the diagonal running from the lower left corner to the upper right corner are common to both the patient progression and survival model. These included metabolites such as hydroxylamine (down-regulated), glucopyranose, tagatose, glutamine, tyramine, and proline (up-regulated). Metabolites such as phosphoric acid, glycine and octadecanoic acid were unique to the disease survival model. Our results thus indicate that a unique biomarker profile is possible which distinguishes progression from survival.
3.3.2 NMR metabolic profiles facilitates evaluation of cancer staging and cancer type from pre-treatment samples
Preliminary modeling of clinical covariates with quantitative NMR-derived metabolic profiles indicated that cancer stage and type were well reflected in the serum profiles. We were able to facilitate discrimination in cancer staging between cancer stages 1 and 2, versus stage 3 using 1H-NMR metabolomic data (Supplemental Fig. 3A) (N = 24, CV-ANOVA p < 0.05, R2 = 0.474, Q2 = 0.314). This was based on 8 metabolites such as 2-hydroxybutyrate, 2-oxoisocaproate, acetate, carnitine, 3-hydroxyisovalerate, 2-hydroxyisovalerate, glycerol and glycine. Summary of the scores from this analysis (Fig. 3a) in which the distribution is plotted according to the class i.e. stages 1 and 2, versus stage 3; patients who were staged lower had a lower score in comparison to patients with higher staging.

NMR metabolite bioprofiling facilitates evaluation of pathological tumor characteristics. a Box and whisker plot reflective of tumor staging: Scores from OPLS-DA analysis after baseline samples were stratified into stages 1 and 2, versus 3; b Box and whisker plot based on cancer cell type with sample stratified as non small cell lung cancer type squamous cell and adenocarcinoma
In a further subgroup analysis, we investigated the metabolomic profiles of non-small cell lung (NSCLC) cancer patients. Using 1H-NMR data we were able to distinguish sera based on cancer pathophysiology. Patients with NSCLC could be discriminated into subtypes of squamous and adenocarcinoma. OPLS-DA modeling was able to discriminate between the two cancer sub-types based on 19 differentially abundant spectral features (Supplemental Fig. 3B) (N = 18, CV-ANOVA p < 0.01, R2 = 0.677, Q2 = 0.536). Figure 3b shows the box and whisker plot plotted using metabolite scores based between two classes of non small cell cancer i.e. squamous cell and adenocarcinoma cell carcinoma respectively. Metabolites such as 2-oxoisocaproate, 4-hydroxybutyrate, lysine, arginine, dimethylamine, isobutyrate, 3-hydroxybutyrate, acetate, asparagine, phenylalanine were relatively higher in patients with adenocarcinoma. However, metabolites such as pyruvate, lactate, valine, proline, isoleucine, histidine, 2-aminobutyrate, leucine and alloisoleucine were relatively lower in the patients with adenocarcinoma.
4 Discussion
This pilot project aimed to examine the feasibility of characterizing metabolomic profiles of lung cancer patients serially over the course of treatment. We analyzed serum specimens collected pre-treatment, at approximately the mid-point of treatment and at 6-months follow-up. While somewhat limited by the sample size of our study, we observed that the metabolite profiles were clearly reflective of both temporal and pathophysiological parameters. Baseline samples naïve to treatment are potentially predictive of crucial clinical parameters such as survival and progression, and reflected tumor pathophysiology (squamous cell vs. adenocarcinoma) and stage (1H-NMR, p < 0.05). These observations are consistent with those made by others (Jordan et al. 2010; Hori et al. 2011). In contrast to other studies, we collected serial samples and observed that the 1H-NMR and GC–MS metabolomics data was reflective of the difference between the various time points at which the samples were collected through the treatment process. Such differences suggest that there may be specific temporal shifts in metabolites during and after which merit further examination.
We also observed that the metabolite profiles appear to correlate with disease progression and survival (GC–MS p < 0.05). While Wedge et al. (2011), observed that particular metabolites were associated with worse survival outcomes among SCLC patients, we examined the overall metabolomic signature rather than identifying and quantifying individual metabolites. Since lung cancer is an entity involving differential metabolic changes in various pathways, the combinational approach of biomarker discovery in the form of a profile could reflect the pathological dynamics of the disease in a more comprehensive fashion.
One possible explanation for the segregation of metabolite pools between those prognostic for survival and those indicative of progression may lie in compartmentalization of biochemical processing and possible mitochondrial dysfunction. Metabolites from our study related to survival, such as tridecan-1-ol and octadecan-1-ol are long chain fatty alcohols which may result from oxidative processing in peroxisomes or from dietary sources. These are ketogenic substrates which are lower in deuterium content (105–130 ppm) than cytosolic water (~155 ppm) (Duan et al. 2002; Schmidt et al. 2003). In turn, mitochondrial processing of ketogenic substrates leads to generation of so-called ‘metabolic’ or ‘matrix’ water (i.e. water generated in the mitochondria) via beta-oxidation (Boros and Somlyai 2015). This metabolic water is consequently relatively depleted in deuterium, and this pool is ultimately used for mitochondrial NADPH dependent macromolecular synthesis, including DNA. By this theory, hydrogen bonding in DNA will be differentially impacted in patients who use ketogenic substrates with lower deuterium content due to isotope effects (Boros et al. 2014; Sobczyk et al. 2013). Conversely, pentose-cycle derived NADPH will have a relatively elevated deuterium content and thus elevation in markers from these non-ketogenic substrates correlated with decreased survival. Elevated circulating fatty alcohols as observed here in patients improved survival characteristics may serve as a marker of increased deuterium depletion from ketogenic substrates, possibly due to differing mitochondrial function. In fact, deuterium depleted water has been shown to inhibit lung tumor growth in vivo as well as reduce proliferation of A549 cell lines with a concomitant increase in apoptosis (Cong 2010). In contrast, tagatose, hydroxylamine, glucopyranose and threonine are glycogenic substrates were found to be indicative of disease progression and do not impact deuterium loading for DNA stabilization to slow tumor growth. The analytical platform dependence of our results may be a result of an indirect bias in detection of ketogenic metabolites by GC–MS.
Differences between our study and other metabolomic studies in lung cancer could relate to variations in sample collection. In the present study, we used serum instead of plasma for our analyses. Previous studies (Yu et al. 2011; Liu et al. 2010; Denery et al. 2011) have indicated that in contrast to plasma, serum demonstrates a higher concentration of metabolites and reduced background noise making it suitable for biomarker discovery. We were not able to control for external environmental factors that may be confounding such as for diurnal variation, diet and smoking status (Carrola et al. 2011; Psihogios et al. 2007). However, comparison of changes in individual patients’ metabolomic profile over time should minimize the influence of intrinsic factors (gender, age, co-morbid medical conditions).
Our study was also unique in that we utilized both complementary technique of 1H-NMR and GC–MS; a similar approach has been utilized by Zhang et al. (2012) and Gu et al. (2011). The sample size studied limits our ability to draw any conclusion about whether 1H-NMR or GC–MS might be the better tool as a whole, although our finding point to each technology having stronger assessments of different clinical parameters. Other groups have used various analytic platforms including 1H-NMR (Carrola et al. 2011; Rocha et al. 2011), GC–MS (Hori et al. 2011; Wedge et al. 2011) and LC–MS (Wen et al. 2013). Each technique affords different advantages and disadvantages. Whereas NMR requires less sample and sample preparation, it is less sensitive and requires more expensive instrumentation. Ultimately, no single analytical technique can likely identify entire range of metabolites present in biological samples and the combination of data from multiple analytical platforms may be complementary.
In conclusion, it is feasible to characterize metabolomic profiles from serum samples of lung cancer patients over the course of their treatment. The relative heterogeneity of the patients, treatments and sample collection in our small pilot study limits our ability to draw any definitive conclusions. Therefore our findings should be interpreted as preliminary and hypothesis generating. Nevertheless, despite the small sample size, our preliminary findings suggest there is variability in the metabolomic profile of lung cancer patients that is associated with staging, prognosis, and survival. Future studies are planned examining a larger, more homogenous group of lung cancer patients whose serial serum samples were prospectively collected as part of a controlled clinical trial. With a larger sample size we hope to more clearly characterize baseline metabolomic profiles as well as any temporal changes in metabolic patterns that occur over the course of treatment.
References
Bathe, O. F., et al. (2011). Feasibility of identifying pancreatic cancer based on serum metabolomics. Cancer Epidemiology, Biomarkers and Prevention, 20(1), 140–147.
Boros, L.G. & Somlyai, G. (2015). Compartmentalized NADPH Synthesis, Intramolecular Deuterium Disequilibrium and Water Pools of Mammalian Cells.
Boros, L. G., et al. (2014). Abstract 1426: Fumarate hydratase and deuterium depletion control oncogenesis via NADPH-dependent reductive synthesis: mitochondrial matrix water, DNA deuteration and epigenetic events. Cancer Research, 74(19 Supplement), 1426.
Carrola, J., et al. (2011). Metabolic signatures of lung cancer in biofluids: NMR-based metabonomics of urine. Journal of Proteome Research, 10(1), 221–230.
Chen, Y., et al. (2009). RRLC-MS/MS-based metabonomics combined with in-depth analysis of metabolic correlation network: Finding potential biomarkers for breast cancer. Analyst, 134(10), 2003–2011.
Cong, F. S. (2010). Deuterium-depleted water inhibits human lung carcinoma cell growth by apoptosis. Experimental and Therapeutic Medicine, 1(2), 277–283.
Deja, S., et al. (2014). Metabolomics provide new insights on lung cancer staging and discrimination from chronic obstructive pulmonary disease. Journal of Pharmaceutical and Biomedical Analysis, 100C, 369–380.
Denery, J. R., Nunes, A. A. K., & Dickerson, T. J. (2011). Characterization of differences between blood sample matrices in untargeted metabolomics. Analytical Chemistry, 83(3), 1040–1047.
Duan, J.-R., et al. (2002). Natural deuterium distribution in fatty acids isolated from peanut seed oil: A site-specific study by quantitative 2H NMR spectroscopy. ChemBioChem, 3(8), 752.
Fan, T. W., et al. (2009). Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Molecular Cancer, 8(1), 41.
Farshidfar, F., et al. (2012). Serum metabolomic profile as a means to distinguish stage of colorectal cancer. Genome medicine, 4(5), 42.
Gu, H., et al. (2011). Principal component directed partial least squares analysis for combining nuclear magnetic resonance and mass spectrometry data in metabolomics: Application to the detection of breast cancer. Analytica Chimica Acta, 686(1–2), 57–63.
Hiller, K., et al. (2009). MetaboliteDetector: Comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Analytical Chemistry, 81(9), 3429–3439.
Hori, S., et al. (2011). A metabolomic approach to lung cancer. Lung Cancer (Amsterdam, Netherlands), 74(2), 284–292.
Jordan, K. W., et al. (2010). Comparison of squamous cell carcinoma and adenocarcinoma of the lung by metabolomic analysis of tissue–serum pairs. Lung Cancer (Amsterdam, Netherlands), 68(1), 44–50.
Liu, L., et al. (2010). Differences in metabolite profile between blood plasma and serum. Analytical Biochemistry, 406(2), 105–112.
Lokhov, P. G., et al. (2013). Blood plasma metabolites and the risk of developing lung cancer in Russia. European Journal of Cancer Prevention, 22(4), 335–341.
Morvan, D., & Demidem, A. (2007). Metabolomics by proton nuclear magnetic resonance spectroscopy of the response to chloroethylnitrosourea reveals drug efficacy and tumor adaptive metabolic pathways. Cancer Research, 67(5), 2150–2159.
Odunsi, K., et al. (2005). Detection of epithelial ovarian cancer using 1H-NMR-based metabonomics. International Journal of Cancer, 113(5), 782–788.
Psihogios, N. G., et al. (2007). Gender-related and age-related urinalysis of healthy subjects by NMR-based metabonomics. NMR in Biomedicine, 21(3), 195–207.
Rocha, C. M., et al. (2011). Metabolic signatures of lung cancer in biofluids: NMR-based metabonomics of blood plasma. Journal of Proteome Research, 10(9), 4314–4324.
Sawyers, C. L. (2008). The cancer biomarker problem. Nature, 452(7187), 548–552.
Schmidt, H. L., Werner, R. A., & Eisenreich, W. (2003). Systematics of 2H patterns in natural compounds and its importance for the elucidation of biosynthetic pathways. Phytochemistry Reviews, 2, 61–85.
Sobczyk, L., Obrzud, M., & Filarowski, A. (2013). H/D isotope effects in hydrogen bonded systems. Molecules, 18(4), 4467–4476.
Sreekumar, A., et al. (2009). Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature, 457(7231), 910–914.
Wedge, D. C., et al. (2011). Is serum or plasma more appropriate for intersubject comparisons in metabolomic studies? An assessment in patients with small-cell lung cancer. Analytical Chemistry, 83(17), 6689–6697.
Weljie, A. M., et al. (2006). Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Analytical Chemistry, 78(13), 4430–4442.
Weljie, A. M., et al. (2007). An inflammatory arthritis-associated metabolite biomarker pattern revealed by 1H NMR spectroscopy. Journal of Proteome Research, 6(9), 3456–3464.
Wen, T., et al. (2013). Exploratory investigation of plasma metabolomics in human lung adenocarcinoma. Molecular BioSystems, 9(9), 2370–2378.
Yang, Q., et al. (2010). Urinary metabonomic study of lung cancer by a fully automatic hyphenated hydrophilic interaction/RPLC-MS system. Journal of Separation Science, 33(10), 1495–1503.
Yu, Z., et al. (2011). Differences between human plasma and serum metabolite profiles. PLoS One, 6(7), e21230.
Zhang, S. (2007). A comprehensive evaluation of SAM, the SAM R-package and a simple modification to improve its performance. BMC Bioinformatics, 8(1), 230.
Zhang, J., et al. (2012). Esophageal cancer metabolite biomarkers detected by LC-MS and NMR methods. PLoS One, 7(1), e30181.
Acknowledgments
This research was supported in part by the Alberta Innovation Health Solutions (AMW) and the Alberta Cancer Board, Alberta Cancer Foundation and the University of Calgary.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare that they have no potential conflicts of interest to disclose.
Ethical approval
This study was approved by the University of Calgary Conjoint Health Research Ethics Board. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Desirée Hao and M. Omair Sarfaraz have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hao, D., Sarfaraz, M.O., Farshidfar, F. et al. Temporal characterization of serum metabolite signatures in lung cancer patients undergoing treatment. Metabolomics 12, 58 (2016). https://doi.org/10.1007/s11306-016-0961-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-016-0961-5