
Abstract
The neurotrophin brain-derived neurotrophic factor (BDNF), which acts as a transducer, is responsible for improving cerebral stroke, neuropathic pain, and depression. Exercise can alter extracellular nucleotide levels and purinergic receptors in central nervous system (CNS) structures. This inevitably activates or inhibits the expression of BDNF via purinergic receptors, particularly the P2X receptor (P2XR), to alleviate pathological progression. In addition, the significant involvement of sensitive P2X4R in mediating increased BDNF and p38-MAPK for intracerebral hemorrhage and pain hypersensitivity has been reported. Moreover, archetypal P2X7R blockade induces mouse antidepressant-like behavior and analgesia by BDNF release. This review summarizes BDNF-mediated neural effects via purinergic receptors, speculates that P2X4R and P2X7R could be priming molecules in exercise-mediated changes in BDNF, and provides strategies for the protective mechanism of exercise in neurogenic disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The neurotrophin brain-derived neurotrophic factor (BDNF) is a key mediator of neuroplastic changes induced by exercise. Physical activity has demonstrated positive effects, such as increased adult neurogenesis, in preventing and ameliorating a wide range of brain diseases. Disquietingly, it is possible to affirm that 46.6% of Parkinson’s disease (PD) patients in the Netherlands were less physically active since the COVID-19 lockdown, and reduced physical activity correlated with exacerbating PD symptoms (rigidity, fatigue, tremor, pain, and concentration) [1]. Encouraging or prescribing regular exercise can be worthwhile, due to major depressive disorder symptoms of low energy and motivation [2]. Meanwhile, adult-born neurons can migrate to regions of damage and facilitate repair with stroke, which can be further enhanced by exercise interventions [3]. In addition, repeated high-intensity swimming exercise reduces mechanical allodynia in complex regional pain syndrome type I mice [4]. Synthesis of BDNF is already the exact mechanism underlying exercise-induced neuroprotection in the hippocampus, hypothalamus, and cortex. This process plays crucial roles in gliogenesis, neurogenesis, synaptogenesis, and angiogenesis [5], leading to the enhancement of brain function [6]. The binding of BDNF to high-affinity tyrosine receptor kinase B (TrkB) can initiate at least 3 intracellular signaling pathways, including the mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MAPK/ERK), phospholipase Cg (PLCγ), and phosphoinositide 3-kinase (PI3K) pathways [7, 8]. Regular exercise stimulated the expression of BDNF and TrkB in a stroke rat model, which can be negated by treatment with antisense BDNF oligonucleotide [9]. Treadmill exercise training increased BDNF and decreased Akt activity in the paraventricular nucleus post-myocardial infarction [10] and protected against IFN-α-induced decreases in the expression of BDNF in the hippocampus and prefrontal cortex [11]. In addition, treatment with a phosphoinositide-13 (PI3) kinase inhibitor reversed the beneficial effects of exercise-induced expression of BDNF in neurorepair [12].
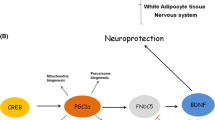
More evidence is still needed to clearly define the exercise-related BDNF mechanisms. On the other hand, the evidence supporting a role for exercise in purines has remained ambiguous. ATP was not only thought to be solely the universal energy currency but also in the nervous system is released into the extracellular space by neurons, astrocytes, and microglia through Panx1 channels to mediate inflammation and glial cell proliferation, which is engaged in the development of diseases such as stroke, epilepsy, and chronic pain [13]. Purinergic receptors (A1, A2A, A2B, P2X4, P2X7, P2Y11Rs) have been recognized as important mediators of BDNF activation and participate in multiple pathologies, including stroke, neuropathic pain, and depression (Table 1). Moreover, preconditioning exercise decreased P2X4 receptor (P2X4R) and BDNF, decreased inflammatory cytokines, and ceased prior to sciatic injury [14]. It has been reported that P2X7 receptor (P2X7R) and BDNF are probably involved in neuron structural and functional modifications that culminate in the enhancement of sensorimotor function after exercise [15]. Here, we provide support for the hypothesis that P2X4R and P2X7R activate the BDNF signaling pathway in the neuroprotection of exercise (Figure 1), which mainly contributes to anti-inflammatory and neuroplasticity.

Hypothesis: Exercise could decrease the expression of BDNF by inhibiting P2X4R activation; exercise could increase the expression of BDNF by enhancing P2X7R activation. Exercise ameliorates neuropathic pain and stroke by regulating P2X4R and P2X7R activation. Eventually, P2X4R and P2X7R may complement neural effects due to the release of BDNF exercise
Exercise improves neurological disorders by regulating BDNF and purinergic signaling
Physical exercise is a well-known and established method for the prevention and treatment of many diseases (metabolic syndrome, hypertension, neuronal and psychiatric disorders, among others) recognized by the American College of Sports Medicine (ACSM) [27,28,29]. Importantly, this includes powerful effects of endurance exercise on the brain and nervous system. As a striking example, a trial of endurance exercise training (moderate-intensity treadmill walking 3 days/week) was conducted in adults 55–80 years of age [30]. Participants in the exercise group exhibited a 2% increase in hippocampal volume that positively correlated with improvements in spatial memory. Moreover, running has been shown to fortify the organization of and communication within neuronal networks, which could have beneficial impacts on memory and spatial-temporal information processing [31]. Importantly, exercise-induced neuronal adaptations are accompanied by the proliferation of endothelial cells and angiogenesis in the cerebral cortex and hippocampus [32]. One mechanism through which acute/chronic physical exercise triggers responses/adaptations locally and globally is through the production and secretion of BDNF or other circulating factors, which beneficially upregulates stress-response pathways, induces vascularization, and promotes synaptic plasticity and neurogenesis [22, 33].
A group of researchers investigated the role of physical exercise in preventing and treating diseases and its relationship with purinergic signaling. Studies were carried out to investigate the modulation of the purinergic system (receptors, enzymes, and nucleotides) by physical exercise. Submitting rats to 20 min of a daily moderate treadmill for 2 weeks can significantly reduce adenosine triphosphate (ATP) and adenosine diphosphate (ADP) hydrolysis in the hippocampal synaptosomes and blood serum [23], suggesting lower activity of CD39. Possible modulation of A1R [4], A2AR, P2X2R, P2X6R [16], P2X4R [14], and P2X7R [15] by physical exercise was explored in complex regional pain syndrome type I (CRPS-I) mice, chronic constriction injury rats, stroke rats, and hypertensive rats. However, there is still a lack of information about the possible effects of physical training on some P2YRs.
Purinergic receptors regulate BDNF
BDNF has been described to increase neuronal excitability and synaptic plasticity [17, 18], whereas the precursor of BDNF, proBDNF, preferentially binds to p75NTR and causes diametrically opposing effects, triggering apoptosis and synaptic depression [19, 20]. BDNF could promote the survival of serotonergic axons, enhancing 5‐HT uptake and its activity-dependent release. In mouse and postmortem sample patients, downregulation of BDNF was related to depression in various brain regions, including the anterior cingulate cortex, caudal brainstem, ventral prefrontal cortex, and hippocampus [21]. Intravenous BDNF injection after stroke could enhance neuronal remodeling, leading to improved functional motor recovery (rotarod, beam balance, neuroscore) [24]. The processes of sustaining chronic pain are also correlated with the sustained release of BDNF, while the maintenance of hyperalgesia has not been completely elucidated [26]. Emerging evidence indicates that A1, A2, and P2Rs are crucial regulatory factors in BDNF expression, which is essential for the release of glutamate vesicles and even microglial reactivity (Table 1). P2X4R and P2X7R are involved in neuropathic pain [34], intracerebral hemorrhage [35], and depression [36] by regulating BDNF actions.
It is accepted that P1 and P2Rs induce changes in BDNF expression in relevant CNS structures. A2AR activation is a determinant of the inhibitory actions of BDNF on GABA release and Glu release [37], the expansionary effects of BDNF on axonal elongation/dendritic branching [38] and the facilitation of LTP on hippocampal CA1 [39]. Hippocampal A1R or A2AR activation could elicit similar elevations in pERK1/2 in a model of global cerebral ischemia reperfusion injury, along with decreased microglial reactivity and increased BDNF expression via A1R [40]. However, a significant increase in BDNF levels was detected after caffeine application (a selective A1R antagonist) in hippocampal slices [41]. In vitro studies have also confirmed that BDNF production is dependent on P2X4R using an ATP-activated spontaneously immortalized microglial cell line [42]. Genetic deletion of P2X7R appears to be the cause of the increase in BDNF [43]. ATP exocytosis induced by the antidepressant fluoxetine increased the astrocytic expression of BDNF mRNA in primary cultures of hippocampal astrocytes, which was mediated by activation of P2Y11R and A2BR. Moreover, intracellular signaling cascades of BDNF release are associated with the accumulation of cAMP and the activation of protein kinase A (PKA), not Ca2+/calmodulin-dependent kinase (CaM kinase) [44]. However, some evidence indicated that ATP-triggered vesicle fusion and BDNF secretion were probably related to astrocytic Ca2+ excitability [45].
Exercise alleviates neuropathic pain by inhibiting the P2X4R-BDNF pathway
Typically, neuropathic pain is characterized by hyperexcitability of the primary afferent sensory neurons accompanied by the release of transmitters or mediators such as BDNF [46] and glutamate [47]. The increased mBDNF/proBDNF ratio mediated by ATP administration was dependent on P2X4R, which was confirmed by P2X4R-shRNA treatment [35]. The communication of microglial P2X4R and neurons is an essential link in pain transmission, and BDNF-TrkB signaling also plays a crucial role in analgesia. Notably, the upregulation of excitatory amino acid transporter 3 (EAAT3), a subtype of sodium-dependent EAATs, was accompanied by increased P2X4R and BDNF in the microglia of rats following trigeminal allodynia induced by repeated dural IS infusions [34]. Moreover, the activated P2X4R in microglia causes the phosphorylation of p38-MAPK, resulting in the release of BDNF, all of which are essential to the persistence of pain hypersensitivity after nerve injury. Therefore, it seems plausible that blockade of the P2X4R-p-p38-MAPK-BDNF pathway in the spinal cord may provide a novel therapeutic strategy for neuropathic pain [25].
Growing evidence suggests that exercise alleviates neuropathic pain in rats. Six weeks of voluntary rotation before the onset of chronic compression injury prevented the full development of ectopic pain, normalized the expression of excitatory interleukin (IL)-1β, and decreased the expression of the P2X4R-BDNF axis in the dorsal spinal cord of the ipsilateral spinal cord [14]. High-intensity exercise caused a significant adenosine concentration in the entire rat brain. In the subsequent sleep period, adenosine levels decrease, and the concentrations of ADP and ATP increase [48]. Low and high exercise frequencies both significantly prevented A2AR overactivation [49]. The changes in purines after exercise observed in experiments support that ATP and nucleotides are able to interact with BDNF to amplify or modulate their signaling, resulting in amelioration of neuropathology.
P2X4R expression in microglia and the subsequent release of BDNF are both required for hyperalgesia. Specifically, P2X4R activation by ATP at the cell surface leads to Ca2+ influx, phosphorylation of p38, and synthesis and release of BDNF, causing disinhibition of nociceptive projection neurons [50]. When P2X4R/Trk-Fc was blocked, inhibition of HSV-1-induced allodynia was triggered [51]. Basically, BDNF can protect neurons against various insults [9, 10] and the neuroprotection of P2X4R after stroke by promoting the synthesis of mBDNF [35]. Moreover, P2X4R upregulation does not occur based on any exercise modalities. Interestingly, preconditioning exercise decreased P2X4R and BDNF, ceased prior to sciatic injury, and decreased neuroimmune signaling in the spinal dorsal horn [14]. Furthermore, in relation to other P2XRs, a high ATP concentration does not activate P2X4R until the pH increases to 7.4 [51]. Therefore, it is highly likely that P2X4R will not be activated to mediate BDNF synthesis during regular or vigorous exercise because H+ production has traditionally been explained by the exercise-induced increased production of lactic acid [52].
Exercise improves stroke probably by activating P2X7R and BDNF
Brain expression of P2X7R has been described in all intrinsic cells of the CNS, and functional P2X7R and downstream signaling pathways are extremely debated. Previous observations have described that longer C-terminal domains of P2X7R are responsible for the activation of downstream signaling pathways, including the ERK pathway and activation of caspase-3, resulting in the initiation of apoptosis [53, 54]. Generally, P2X7R has detrimental and beneficial roles in the nervous system. On the one hand, evidence strongly indicates that excessive P2X7R activation is involved in the caspase-independent death of neural progenitor cells [55]. In particular, prolonged activation of P2X7R is thought to be largely associated with pathological conditions where the extracellular ATP concentration rises dramatically due to inflammation or leakage from cell damage [56]. Accordingly, the involvement of P2X7R during the pathogenesis of multiple chronic disorders of the CNS has been demonstrated [57, 58]. On the other hand, P2X7R stimulation with low amounts of Bz-ATP in rat embryonic NPCs induced neuronal differentiation rather than cell death [59]. Meanwhile, BDNF plays a crucial role in the development, differentiation, and survival of neuronal populations within the central and peripheral nervous systems. In relation to inflammation, there is no definite mechanism for the effects of proinflammatory cytokines on the expression of BDNF, whereas proinflammatory cytokines result in a significant reduction in BDNF [60].
Traditionally, the expression of P2X7 is negatively correlated with BDNF. The P2X7R antagonist A-804598 (3, 10, or 30 mg/kg/day) was used to investigate BDNF signaling in the rat frontal cortex and ventral and dorsal hippocampus. The results showed that antagonizing P2X7R may activate TrkB and mediate an increase in BDNF-AKT-p70 S6 kinase levels in the ventral hippocampus, which produces a related antidepressant effect [36]. Elevated basal BDNF levels and neurogenesis in the hippocampus of P2X7R−/− mice compared with P2X7R+/+ controls indicate tonic inhibitory regulation of BDNF production through P2X7R [43]. In line with this argument, experimental models have demonstrated that inhibition of P2X7R alleviates depression via BDNF activation [36]. Unfortunately, pharmacological antagonists of P2X7R have been produced and tested in various diseases with mostly disappointing results [61, 62]. Another study, however, suggests otherwise. Central poststroke pain (CPSP) was used to examine continuous sensitization behavior in rats, which was suppressed by a P2X7R antagonist and BDNF receptor blocker. The results demonstrated that treatment of stroke pain with a P2X7R antagonist can prevent microglial P2X7R activation in the surrounding areas of CPSP lesions and reduce the release of regional inflammatory cytokines [63]. More importantly, Glu release is an important function of P2X7R activation related to BDNF expression; in turn, subsequent overactivation of extrasynaptic NR2B receptors (NMDA receptor subunits) downregulates BDNF expression [43]. It has been reported that P2X7R controls BDNF release from Schwann cells, which exert neuroprotective effects on neighboring vestibular neurons [64]. Therefore, negative feedback loops of molecular interaction (P2X7R and BDNF) in cells may be the main mechanism of disease and therapy. In addition, considering that P2X7R cooperates with NMDA and BDNF receptors to promote neuronal survival through both the PKC and PI3K/Akt pathways, a precise interpretation of the results is that P2X7R and BDNF play neuroprotective roles.
Interestingly, preconditioning exercise significantly reduced infarct volume and apoptosis, in which P2X7R and BDNF in the ischemic brain were significantly upregulated. Following intracerebral hemorrhage, ATP administration had the ability to relieve cerebral hemorrhage–induced injury and improve cerebral neurological function by upregulating the mBDNF/proBDNF ratio and p38-MAPK. However, whether P2X7R has a clear regulatory effect on BDNF has not been clearly demonstrated in the area of stroke improvement [15].
Conclusion
Owing to the relatively few discussions of the purinergic constituents in neuroprotection during or after exercise, it is pressing to explore their physiology. As discussed in this review, some P1/P2Rs have been recognized as significant mediators of BDNF expression, which participate in multiple pathologies, including stroke, neuropathic pain, and even depression. Both P2X4R and P2X7R influence BDNF activation, leading to accumulation of p38-MAPK and upregulation of Iba1 and pERK1/2. A1, A2A, A2B, P2X4, P2X7, and P2Y11Rs have been shown to participate in regulating BDNF expression. However, interactions between the downstream signaling mechanisms of P2X7R may also occur. Eventually, P2X4R and P2X7R may complement neural effects due to the release of BDNF by exercise.
Data availability
Not applicable.
Code availability
Not applicable.
Abbreviations
- ATP:
-
adenosine triphosphate
- ADP:
-
adenosine diphosphate
- CNS:
-
central nervous system
- TrkB:
-
tyrosine receptor kinase B
- MAPK:
-
mitogen-activated protein kinase
- ERK:
-
extracellular signal-regulated protein kinase
- PI3K:
-
phosphoinositide 3-kinase
- PLCγ:
-
phospholipase Cg
- EAAT3:
-
excitatory amino acid transporter 3
- CPSP:
-
central post stroke pain
- PD:
-
Parkinson’s disease
- Glu:
-
glutamic acid
- GABA:
-
gamma-aminobutyric acid
- —R:
-
receptor
References
Brown EG, Chahine LM, Goldman SM, Korell M, Mann E, Kinel DR, Arnedo V, Marek KL, Tanner CM (2020) The effect of the COVID-19 pandemic on people with Parkinson’s disease. J Parkinson’s Dis 10(4):1365–1377. https://doi.org/10.3233/JPD-202249
Köhler-Forsberg O, Cusin C, Nierenberg AA (2019) Evolving issues in the treatment of depression. JAMA 321:2401–2402. https://doi.org/10.1001/jama.2019.4990
Xiong Y, Mahmood A, Chopp M (2009) Emerging treatments for traumatic brain injury. Expert Opin Emerg Drugs 14:67–84. https://doi.org/10.1517/14728210902769601
Martins DF, Mazzardo-Martins L, Soldi F, Stramosk J, Piovezan AP, Santos AR (2013) High-intensity swimming exercise reduces neuropathic pain in an animal model of complex regional pain syndrome type I: evidence for a role of the adenosinergic system. Neuroscience 234:69–76. https://doi.org/10.1016/j.neuroscience.2012.12.042
Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S (2008) New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev 59:201–220. https://doi.org/10.1016/j.brainresrev.2008.07.007
El-Sayes J, Harasym D, Turco CV, Locke MB, Nelson AJ (2019) Exercise-induced neuroplasticity: a mechanistic model and prospects for promoting plasticity. Neuroscientist : Rev J Bring Neurobiol, Neurol Psych 25:65–85. https://doi.org/10.1177/1073858418771538
Schäbitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S (2000) Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke 31:2212–2217. https://doi.org/10.1161/01.str.31.9.2212
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225. https://doi.org/10.1016/s0092-8674(00)00114-8
Sakakima H, Khan M, Dhammu TS, Shunmugavel A, Yoshida Y, Singh I, Singh AK (2012) Stimulation of functional recovery via the mechanisms of neurorepair by S-nitrosoglutathione and motor exercise in a rat model of transient cerebral ischemia and reperfusion. Restor Neurol Neurosci 30:383–396. https://doi.org/10.3233/RNN-2012-110209
Lee HW, Ahmad M, Wang HW, Leenen F (2020) Effects of exercise on BDNF-TrkB signaling in the paraventricular nucleus and rostral ventrolateral medulla in rats post myocardial infarction. Neuropeptides 82:102058. https://doi.org/10.1016/j.npep.2020.102058
Callaghan CK, Rouine J, O’Mara SM (2017) Exercise prevents IFN-α-induced mood and cognitive dysfunction and increases BDNF expression in the rat. Physiol Behav 179:377–383. https://doi.org/10.1016/j.physbeh.2017.07.018
Chen MJ, Russo-Neustadt AA (2005) Exercise activates the phosphatidylinositol 3-kinase pathway. Brain Res Mol Brain Res 135:181–193. https://doi.org/10.1016/j.molbrainres.2004.12.001
Yeung AK, Patil CS, Jackson MF (2020) Pannexin-1 in the CNS: emerging concepts in health and disease. J Neurochem 154:468–485. https://doi.org/10.1111/jnc.15004
Grace PM, Fabisiak TJ, Green-Fulgham SM, Anderson ND, Strand KA, Kwilasz AJ, Galer EL, Walker FR, Greenwood BN, Maier SF, Fleshner M, Watkins LR (2016) Prior voluntary wheel running attenuates neuropathic pain. Pain 157:2012–2023. https://doi.org/10.1097/j.pain.0000000000000607
Otsuka S, Sakakima H, Tani A, Nakanishi K, Takada S, Norimatsu K, Maejima H, Maruyama I (2021) Effects of detraining on preconditioning exercise-induced neuroprotective potential after ischemic stroke in rats. Brain Struct Funct 226:2169–2180. https://doi.org/10.1007/s00429-021-02317-5
Cardoso AM, Manfredi LH, Zanini D, Bagatini MD, Gutierres JM, Carvalho F, Tremblay A, Belló-Klein A, Rubin MA, Morsch VM, Sévigny J, Schetinger M (2019) Physical exercise prevents memory impairment in an animal model of hypertension through modulation of CD39 and CD73 activities and A2A receptor expression. J Hypertens 37:135–143. https://doi.org/10.1097/HJH.0000000000001845
Amaral MD, Pozzo-Miller L (2007) BDNF induces calcium elevations associated with IBDNF, a nonselective cationic current mediated by TRPC channels. J Neurophysiol 98:2476–2482. https://doi.org/10.1152/jn.00797.2007
Blum R, Kafitz KW, Konnerth A (2002) Neurotrophin-evoked depolarization requires the sodium channel Na(V)1.9. Nature 419:687–693. https://doi.org/10.1038/nature01085
Gibon J, Buckley SM, Unsain N, Kaartinen V, Séguéla P, Barker PA (2015) proBDNF and p75NTR control excitability and persistent firing of cortical pyramidal neurons. J Neurosci 35:9741–9753. https://doi.org/10.1523/JNEUROSCI.4655-14.2015
Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14:7–23. https://doi.org/10.1038/nrn3379
Zhang E, Liao P (2020) Brain-derived neurotrophic factor and post-stroke depression. J Neurosci Res 98:537–548. https://doi.org/10.1002/jnr.24510
Cotman CW, Berchtold NC (2002) Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci 25:295–301. https://doi.org/10.1016/s0166-2236(02)02143-4
Siqueira IR, Elsner VR, Rilho LS, Bahlis MG, Bertoldi K, Rozisky JR, Batasttini AM, Torres IL (2010) A neuroprotective exercise protocol reduces the adenine nucleotide hydrolysis in hippocampal synaptosomes and serum of rats. Brain Res 1316:173–180. https://doi.org/10.1016/j.brainres.2009.11.076
Schäbitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, Schwab S, Sommer C (2004) Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke 35:992–997. https://doi.org/10.1161/01.STR.0000119754.85848.0D
Trang T, Beggs S, Wan X, Salter MW (2009) P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci 29:3518–3528. https://doi.org/10.1523/JNEUROSCI.5714-08.2009
Cappoli N, Tabolacci E, Aceto P, Dello Russo C (2020) The emerging role of the BDNF-TrkB signaling pathway in the modulation of pain perception. J Neuroimmunol 349:577406. https://doi.org/10.1016/j.jneuroim.2020.577406
Lammers MD, Anéli NM, de Oliveira GG, de Oliveira Maciel S, Zanini D, Mânica A, de Resende E, Silva DT, Bagatini MD, Sévigny J, De Sá CA, Manfredi LH, Cardoso AM (2020) The anti-inflammatory effect of resistance training in hypertensive women: the role of purinergic signaling. J Hypertens 38(12):2490–2500. https://doi.org/10.1097/HJH.0000000000002578
Pedersen BK, Saltin B (2015) Exercise as medicine - evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand J Med Sci Sports 25(Suppl 3):1–72. https://doi.org/10.1111/sms.12581
American College of Sports Medicine (2021) ACMS’s guideline for exercise testing and prescription. Wolters Kluwer, Philadelphia
Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, Kim JS, Heo S, Alves H, White SM, Wojcicki TR, Mailey E, Vieira VJ, Martin SA, Pence BD, Woods JA, McAuley E, Kramer AF (2011) Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U.S.A 108:3017–3022. https://doi.org/10.1073/pnas.1015950108
Vivar C, Peterson BD, van Praag H (2016) Running rewires the neuronal network of adult-born dentate granule cells. NeuroImage 131:29–41. https://doi.org/10.1016/j.neuroimage.2015.11.031
Van der Borght K, Kóbor-Nyakas DE, Klauke K, Eggen BJ, Nyakas C, Van der Zee EA, Meerlo P (2009) Physical exercise leads to rapid adaptations in hippocampal vasculature: temporal dynamics and relationship to cell proliferation and neurogenesis. Hippocampus 19:928–936. https://doi.org/10.1002/hipo.20545
Wang R, Holsinger R (2018) Exercise-induced brain-derived neurotrophic factor expression: therapeutic implications for Alzheimer’s dementia. Ageing Res Rev 48:109–121. https://doi.org/10.1016/j.arr.2018.10.002
Liu C, Zhang Y, Liu Q, Jiang L, Li M, Wang S, Long T, He W, Kong X, Qin G, Chen L, Zhang Y, Zhou J (2018) P2X4-receptor participates in EAAT3 regulation via BDNF-TrkB signaling in a model of trigeminal allodynia. Mol Pain 14:1744806918795930. https://doi.org/10.1177/1744806918795930
He Q, Li Z, Li T, Zhang Z, Zhao J (2021) ATP stimulation promotes functional recovery after intracerebral haemorrhage by increasing the mBDNF/proBDNF ratio. Neuroscience 459:104–117. https://doi.org/10.1016/j.neuroscience.2020.12.034
Ribeiro DE, Müller HK, Elfving B, Eskelund A, Joca SR, Wegener G (2019) Antidepressant-like effect induced by P2X7 receptor blockade in FSL rats is associated with BDNF signalling activation. J Psychopharmacol 33:1436–1446. https://doi.org/10.1177/0269881119872173
Vaz SH, Lérias SR, Parreira S, Diógenes MJ, Sebastião AM (2015) Adenosine A2A receptor activation is determinant for BDNF actions upon GABA and glutamate release from rat hippocampal synaptosomes. Purinergic Signal 11:607–612. https://doi.org/10.1007/s11302-015-9476-1
Ribeiro FF, Neves-Tomé R, Assaife-Lopes N, Santos TE, Silva RF, Brites D, Ribeiro JA, Sousa MM, Sebastião AM (2016) Axonal elongation and dendritic branching is enhanced by adenosine A2A receptors activation in cerebral cortical neurons. Brain Struct Funct 221:2777–2799. https://doi.org/10.1007/s00429-015-1072-1
Jerónimo-Santos A, Batalha VL, Müller CE, Baqi Y, Sebastião AM, Lopes LV, Diógenes MJ (2014) Impact of in vivo chronic blockade of adenosine A2A receptors on the BDNF-mediated facilitation of LTP. Neuropharmacology 83:99–106. https://doi.org/10.1016/j.neuropharm.2014.04.006
Atef RM, Agha AM, Abdel-Rhaman AA, Nassar NN (2018) The ying and yang of adenosine A1 and A2A receptors on ERK1/2 activation in a rat model of global cerebral ischemia reperfusion injury. Mol Neurobiol 55:1284–1298. https://doi.org/10.1007/s12035-017-0401-1
Lao-Peregrín C, Ballesteros JJ, Fernández M, Zamora-Moratalla A, Saavedra A, Gómez Lázaro M, Pérez-Navarro E, Burks D, Martín ED (2017) Caffeine-mediated BDNF release regulates long-term synaptic plasticity through activation of IRS2 signaling. Addict Biol 22:1706–1718. https://doi.org/10.1111/adb.12433
Dave KM, Ali L, Manickam DS (2020) Characterization of the SIM-A9 cell line as a model of activated microglia in the context of neuropathic pain. PLoS One 15:e0231597. https://doi.org/10.1371/journal.pone.0231597
Csölle C, Baranyi M, Zsilla G, Kittel A, Gölöncsér F, Illes P, Papp E, Vizi ES, Sperlágh B (2013) Neurochemical changes in the mouse hippocampus underlying the antidepressant effect of genetic deletion of P2X7 receptors. PLoS One 8:e66547. https://doi.org/10.1371/journal.pone.0066547
Kinoshita M, Hirayama Y, Fujishita K, Shibata K, Shinozaki Y, Shigetomi E, Takeda A, Le H, Hayashi H, Hiasa M, Moriyama Y, Ikenaka K, Tanaka KF, Koizumi S (2018) Anti-depressant fluoxetine reveals its therapeutic effect via astrocytes. EBioMedicine 32:72–83. https://doi.org/10.1016/j.ebiom.2018.05.036
Stenovec M, Lasič E, Božić M, Bobnar ST, Stout RF Jr, Grubišić V, Parpura V, Zorec R (2016) Ketamine inhibits ATP-evoked exocytotic release of brain-derived neurotrophic factor from vesicles in cultured rat astrocytes. Mol Neurobiol 53:6882–6896. https://doi.org/10.1007/s12035-015-9562-y
Wu Y, Shen Z, Xu H, Zhang K, Guo M, Wang F, Li J (2021) BDNF participates in chronic constriction injury-induced neuropathic pain via transcriptionally activating P2X7 in primary sensory neurons. Mol Neurobiol. https://doi.org/10.1007/s12035-021-02410-0.10.1007/s12035-021-02410-0
Gong K, Kung LH, Magni G, Bhargava A, Jasmin L (2014) Increased response to glutamate in small diameter dorsal root ganglion neurons after sciatic nerve injury. PLoS One 9:e95491. https://doi.org/10.1371/journal.pone.0095491
Dworak M, Diel P, Voss S, Hollmann W, Strüder HK (2007) Intense exercise increases adenosine concentrations in rat brain: implications for a homeostatic sleep drive. Neuroscience 150:789–795. https://doi.org/10.1016/j.neuroscience.2007.09.062
Costa MS, Ardais AP, Fioreze GT, Mioranzza S, Botton PH, Portela LV, Souza DO, Porciúncula LO (2012) Treadmill running frequency on anxiety and hippocampal adenosine receptors density in adult and middle-aged rats. Prog Neuropsychopharmacol Biol Psychiatry 36:198–204. https://doi.org/10.1016/j.pnpbp.2011.10.015
Beggs S, Trang T, Salter M (2012) P2X4R+ microglia drive neuropathic pain. Nat Neurosci 15:1068–1073. https://doi.org/10.1038/nn.3155
Suurväli J, Boudinot P, Kanellopoulos J, Rüütel Boudinot S (2017) P2X4: a fast and sensitive purinergic receptor. Biomed J 40:245–256. https://doi.org/10.1016/j.bj.2017.06.010
Kemp G, Böning D, Beneke R, Maassen N (2006) Explaining pH change in exercising muscle: lactic acid, proton consumption, and buffering vs. strong ion difference. Am J Physiol Regul Integr Comp Physiol 291:R235–R239. https://doi.org/10.1152/ajpregu.00662.2005
Smart ML, Gu B, Panchal RG, Wiley J, Cromer B, Williams DA, Petrou S (2003) P2X7 receptor cell surface expression and cytolytic pore formation are regulated by a distal C-terminal region. J Biol Chem 278:8853–8860. https://doi.org/10.1074/jbc.M211094200
Costa-Junior HM, Sarmento Vieira F, Coutinho-Silva R (2011) C terminus of the P2X7 receptor: treasure hunting. Purinergic Signal 7:7–19. https://doi.org/10.1007/s11302-011-9215-1
Delarasse C, Gonnord P, Galante M, Auger R, Daniel H, Motta I, Kanellopoulos JM (2009) Neural progenitor cell death is induced by extracellular ATP via ligation of P2X7 receptor. J Neurochem 109:846–857. https://doi.org/10.1111/j.1471-4159.2009.06008.x
Illes P, Khan TM, Rubini P (2017) Neuronal P2X7 receptors revisited: do they really exist? J Neurosci 37:7049–7062. https://doi.org/10.1523/JNEUROSCI.3103-16.2017
Deussing JM, Arzt E (2018) P2X7 receptor: a potential therapeutic target for depression? Trends Mol Med 24:736–747. https://doi.org/10.1016/j.molmed.2018.07.005
Zhao H, Chen Y, Feng H (2018) P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr Neuropharmacol 16:1282–1295. https://doi.org/10.2174/1570159X16666180516094500
Tsao HK, Chiu PH, Sun SH (2013) PKC-dependent ERK phosphorylation is essential for P2X7 receptor-mediated neuronal differentiation of neural progenitor cells. Cell Death Dis 4:e751. https://doi.org/10.1038/cddis.2013.274
Zhang JC, Yao W, Hashimoto K (2016) Brain-derived neurotrophic factor (BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr Neuropharmacol 14:721–731. https://doi.org/10.2174/1570159x14666160119094646
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S (2017) The P2X7 receptor in infection and inflammation. Immunity 47:15–31. https://doi.org/10.1016/j.immuni.2017.06.020
Young C, Górecki DC (2018) P2RX7 purinoceptor as a therapeutic target-the second coming? Front Chem 6:248. https://doi.org/10.3389/fchem.2018.00248
Kuan YH, Shih HC, Shyu BC (2018) Involvement of P2X7 receptors and BDNF in the pathogenesis of central poststroke pain. Advances in experimental medicine and biology 1099:211–227. https://doi.org/10.1007/978-981-13-1756-9_18
Verderio C, Bianco F, Blanchard MP, Bergami M, Canossa M, Scarfone E, Matteoli M (2006) Cross talk between vestibular neurons and Schwann cells mediates BDNF release and neuronal regeneration. Brain Cell Biol 35:187–201. https://doi.org/10.1007/s11068-007-9011-6
Funding
This work was supported by the National Key Research and Development Programs of China (2018YFF0300604, 2019YFF0301704), the National Natural Science Foundation of China (81704190, 82074576), the Science and Technology Projects of Sichuan Province (2019YFS0526) , and the Sichuan Provincial Administration of Traditional Chinese Medicine (2021MS444, 2021MS066).
Author information
Authors and Affiliations
Contributions
Bing-xin Sun is the first author and is responsible for collecting materials and writing the first draft of the manuscript. Ai-shi Peng and Pei-Jie Liu edited the review tables and figures. Liang Kang, Min-jia Wang, and Hai-li Ding are responsible for critical revisions of the article. Funds of this article are supported by Liang Kang, Yu-shi Hu, and Min-jia Wang. All the authors contributed to the manuscript revision and read and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, Bx., Peng, As., Liu, Pj. et al. Neuroprotection of exercise: P2X4R and P2X7R regulate BDNF actions. Purinergic Signalling 19, 297–303 (2023). https://doi.org/10.1007/s11302-022-09879-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-022-09879-x