
Abstract
This review article presents a collection of tool compounds that selectively block and are recommended for studying P2Y and P2X receptor subtypes, investigating their roles in physiology and validating them as future drug targets. Moreover, drug candidates and approved drugs for P2 receptors will be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Membrane receptors activated by extracellular purines are subdivided into different subfamilies, (i) nucleotide-activated P2Y and P2X receptors [1], (ii) adenosine receptors [2, 3], and (iii) adenine-activated receptors [4], which are still poorly explored. Except for the ATP-activated P2X ion channel receptor family, all of the other purine receptors belong to the class of rhodopsin-like G protein-coupled receptors (GPCRs). There is a metabolic link between the agonists for the different receptor families, at least those activated by purines, since their physiological ligands are structurally related and can be interconverted by enzymes. For example, the nucleotide ATP, the main P2X receptor agonist, is hydrolyzed by ectonucleotidases furnishing the nucleoside adenosine which activates adenosine receptors [5].
P2X receptors
The P2X receptors are homo- or hetero-trimeric ATP-gated ion channels [6]. Seven different subunits exist, P2X1-P2X7. Besides the homomers, heteromeric receptors exist, the best investigated one being the P2X2/3 receptor.
P2X receptor agonists
All P2X receptors are activated by ATP (1, see Fig. 1), although with different potencies (see Table 1). The P2X7 receptor is the least sensitive subtype requiring high concentrations for activation, sometimes up to the millimolar range, while the P2X1 receptor is the most sensitive receptor activated by submicromolar concentrations of ATP. The P2X2, P2X3, and P2X4 receptors are activated by low micromolar concentrations. Whereas the P2X1 and P2X3 receptors show fast desensitization, the others are less quickly desensitized, and prolonged activation of the P2X7 receptor even leads to pore formation in the cell membrane. BzATP (2) is frequently used instead of ATP as a more potent P2X receptor agonist. This compound is, however, virtually inactive at the rat P2X4 receptor (see Table 1). Subtype-selective agonists are currently not available.

Structures of the P2X receptor agonist ATP and selected P2X receptor antagonists recommended as pharmacological tools or developed as drugs
P2X receptor antagonists
A frequently used ATP-derived, non-selective competitive P2X receptor antagonist is TNP-ATP (3, see Figs. 1 and 2, and Table 1), which is particularly potent at P2X1 and P2X3 receptors, and an X-ray co-crystal structure with the human P2X3 receptor has been obtained (see below). Selective P2X receptor antagonists have been developed for P2X1, P2X2, P2X3, P2X4, and P2X7 receptors (see Table 1) [6,7,8,9,10].

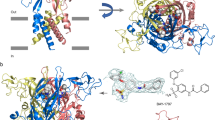
Co-crystal structures of P2X receptors and their ligands, the agonist ATP and a variety of orthosteric and allosteric antagonists. For the P2X1 receptor antagonist, a docked pose is shown. For references see text
P2X1 receptor antagonists
The symmetrical polysulfonated naphthyl derivative NF023 (4, Fig. 1 and Table 1) derived from suramin appears to act as a competitive P2X1 receptor antagonist [11]. Due to its polyanionic character, it is well soluble in water, but its selectivity is limited since it also blocks the P2X3 receptor at somewhat higher concentration. The first relatively potent, selective P2X1 receptor antagonists have recently been described, salicylamide derivatives 5 and 6 [12]. These compounds act as allosteric inhibitors, and their binding site was proposed by docking studies to be located in the extracellular domain (see Fig. 2). They can also inhibit P2X4 and P2X7 receptors at higher concentrations (see Table 1).
P2X2 receptor antagonists
Compound NF770 (7), another symmetrical suramin-derived polysulfonated naphthyl derivative, was described as a potent and relatively selective competitive antagonist of rat P2X2 receptors [13]. However, it also blocks the rat P2X3 receptor at somewhat higher concentration. The Reactive Blue 2 (RB2)-related sulfoanthraquinone derivatives PSB-10211 (8) and PSB-1011 (9) act as potent, but moderately selective rat P2X2 receptor antagonists, probably displaying an allosteric mechanism of inhibition [14]. Recently, ambroxol (10) a bronchosecretolytic drug, which is frequently used to treat bronchial diseases since more than 40 years, was discovered to block the human P2X2 receptor with low micromolar potency [15]. Based on its structure, it likely displays an allosteric mechanism of inhibition. The newly discovered P2X2 receptor blockade might contribute to ambroxol’s biological activity since plasma concentrations should be sufficiently high [16]. Ambroxol likely acts as a multi-target drug also interacting with other targets [17,18,19].
P2X3 receptor antagonists
The P2X3 receptor has been in the focus of drug development efforts by pharmaceutical companies since decades due to their expected analgesic and anti-inflammatory effects [19], and therefore, a number of potent P2X3 receptor antagonists have been developed (Fig. 1) [20,21,22]. The orally administered potent and selective allosteric P2X3 antagonist gefapixant (AF-219, 11) successfully passed a phase III clinical trial for the treatment of refractory chronic cough [23] and is likely to become the first P2X3 receptor antagonist that will be approved as a drug [24]. This has revived the field, and more P2X3 receptor antagonists are now being developed and clinically evaluated for various indications (see, e.g., [21, 25,26,27]). The etymology of the international non-proprietary name (INN) “gefapixant” is explained in Fig. 3 — in fact, it contains part of Geoffrey Burnstock’s name. A co-crystal structure of gefapixant with the human P2X3 receptor has been solved [28]. Its allosteric binding site is located near the orthosteric ATP binding site (see Fig. 2). Structurally related potent P2X3 receptor antagonists include AF-353 (12) and AF-906 (13), all of which are highly potent and perorally bioavailable. The more lipophilic antagonist 12 is even brain-permeant. Antagonists 11–13 also block the heteromeric P2X2/3 receptor subtype at somewhat higher concentrations. In contrast, the imidazopyridine derivative BLU-5937 (14) was reported to be selective for the homomeric P2X3 receptor; it is currently evaluated in clinical trials for the treatment of chronic cough and pruritus [29]. Eliapixant (BAY-181780, 15), another potent and selective P2X3 receptor antagonist [21, 27, 30], has completed a phase 2a clinical trial for the treatment of refractory chronic cough [25] and is further clinically evaluated for the treatment of overactive bladder; another pursued indication is neuropathic pain. A further P2X3 antagonist with undisclosed structure (S-600918) is evaluated in clinical studies [10, 31]. Besides negative allosteric modulators, orthosteric P2X3 receptor antagonists have also been described, e.g., A-317491 (16) [32], which bears three carboxylate functions that interact with basic amino acids in the triphosphate binding pocket as demonstrated by an X-ray co-crystal structure (see Fig. 2) [33].

Naming of the clinically most advanced, most promising P2X receptor antagonist
P2X4 receptor antagonists
5-BDBD (17, Fig. 1 and Table 1), a diazepinone derivative, is a moderately potent and selective allosteric P2X4 receptor antagonist [34]; its main drawback is its low water solubility. A structurally related diazepinedione derivative, NP-1815-PX (18), developed by Japanese researchers around Kazuhide Inoue, is well water-soluble as a sodium salt; but due to its polarity, it does not penetrate into the central nervous system (CNS) [35]. The first P2X4 antagonist, which was evaluated in a clinical trial (phase I), is NC-2600 (19, structure undisclosed) [36]; no further development has been reported despite the positive outcome of the study. A potent, brain-permeable allosteric P2X4 receptor antagonist that is potent at human, rat, and mouse P2X4 receptors and highly selective versus the other P2X receptor subtypes is PSB-15417 (20, structure undisclosed) developed in our group. It showed high efficacy in animal models of neuropathic pain [37]. The N-(benzyloxycarbonyl)phenoxazine derivative PSB-12054 (21) [38] and the urea derivative BX430 (22) [39], moderately potent allosteric P2X receptor antagonists with limited water solubility, are more potent at human than at rodent receptors and can therefore only be recommended for studying the human P2X4 receptor. In contrast, the recently described sulfonamide derivative BAY-1797 (23) [40] is similarly potent at human, rat, and mouse P2X4 receptors showing good P2X4 selectivity but moderate potency. The allosteric antagonist is well water-soluble. Due to its polar character, 23 does not penetrate well into the brain and is only peripherally active.
P2X7 receptor antagonists
The development of P2X7 receptor antagonists is most advanced since the receptor has early been regarded as a promising drug target for treating inflammatory diseases, but the first clinical trials were not successful [10]. Most of the described potent and selective antagonists are negative allosteric modulators, many of which have been optimized with regard to their pharmacokinetic properties [41, 42]. Structurally diverse compounds are available, e.g., 24–28. Antagonist AZ10606120 24 has a particularly high water solubility (25 mM), while compounds 26–28 have been described to penetrate well into the brain. JNJ47965567 (27) and related antagonists were co-crystallized with the human P2X7 receptor and shown to interact with a peripheral binding site far from the orthosteric ATP binding region (see Fig. 2) [43]. JNJ54175446 (28) is a brain-permeable drug that is clinically evaluated for the treatment of major depression and bipolar disorders [44].
Ortho- and allosteric ligand binding sites
The first crystal structure of a P2X receptor in complex with its agonist ATP (Fig. 2), namely, that of the zebrafish P2X4 receptor, confirmed the trimeric structure and the location of the orthosteric binding site within each of the three subunits [45, 46]. ATP was found to bind in an unusual U-shaped conformation (see Fig. 2). Important interactions include (i) polar interactions between the anionic γ-phosphate group of ATP with basic amino acid residues (K72, R298, and K316) and (ii) hydrogen bond interactions of the N6-amino group of ATP with K70 and T189 [47]. Structures of the human P2X3 receptor in complex with the orthosteric antagonists TNP-ATP (3) and A-317491 (16) provided molecular insights in the binding of competitive antagonists [33]. Interestingly, TNP-ATP, a derivative of ATP that is substituted at the ribose-2’- and 3’-hydroxy groups, displays partly similar interactions and binds in a similar position as the agonist ATP. Its additional lipophilic, aromatic trinitrophenyl substituent binds in a deep hydrophobic cleft between two receptor subunits. A very similar binding position is observed for A-317419 (16), in which the carboxylate functions attached to a phenyl ring adopt the role of the nucleotidic phosphate groups. The particular orientation of the lipophilic substituents, present in the antagonists 3 and 16, and their interactions restrict the upward movement of the dorsal fin domain which is required for channel opening.
Besides the orthosteric binding site for ATP, the P2X receptors harbor additional allosteric binding sites which can result in a modulation of the agonist activity (see Fig. 2). The crystal structure of the human P2X3 receptor in complex with the negative allosteric modulator gefapixant (11) revealed its binding between the left flipper and the lower body of the receptor, not too far from the orthosteric site [28]. The inhibitor forms hydrogen bond interactions with the main chain of N190 and the side chain of K176 supported by hydrophobic interactions with V61, V238, and L265 (see Fig. 2). The crystal structures of the human P2X7 receptor with the antagonist JNJ47965567 (27) and four other antagonists identified a common allosteric drug binding site in a groove formed between two adjacent subunits in the upper body domain of the receptor [43]. The allosteric drug binding pocket becomes narrower upon ATP binding, a conformational rearrangement that is crucial for P2X7 receptor channel opening. This action is blocked by antagonists such as 27, which binds deep within the cavity, primarily mediated by hydrophobic interactions with F88, F95, F103, M105, F293, and V312 (see Fig. 2). To provide a structural hypothesis for the recently described series of allosteric P2X1 receptor antagonists, a homology model was generated based on the crystal structures of the P2X7 receptor [12]. The putative binding mode of PSB-2001 (5) and its close analogs partly overlaps with the allosteric binding site identified by the crystal structures of the antagonist-bound P2X7 receptor. The aniline moiety of PSB-2001 is proposed to be embedded forming hydrophobic interactions with F92, F291, F293, F308, and V310, and the salicylate moiety is likely surrounded by another set of hydrophobic residues, F106 and W164 (see Fig. 2) [12].
P2Y receptors
P2Y receptors are G protein-coupled receptors belonging to the δ-branch of the rhodopsin-like receptor family. They are activated by nucleotides such as ATP, ADP, UTP, UDP, or UDP-glucose depending on the receptor subtype (see Fig. 4) [48]. The P2Y1-like receptors include P2Y1 (activated by ADP), P2Y2 (ATP, UTP), P2Y4 (UTP), P2Y6 (UDP), and P2Y11 (ATP). The P2Y12-like receptor family consists of three subtypes, P2Y12 (ADP), P2Y13 (ADP), and P2Y14 (UDP, UDP-glucose). P2Y1, P2Y2, P2Y4, and P2Y6 couple to Gq proteins leading to the activation of phospholipase C, release of inositol trisphosphate (IP3), and intracellular calcium mobilization. The P2Y11 receptor is only present in primates; it can activate phospholipase C via Gq proteins, as well as adenylate cyclase via Gs proteins. P2Y12, P2Y13, and P2Y14 receptors inhibit adenylate cyclase via Gi proteins (see Fig. 4). Some of the receptors can also be activated by dinucleotides, e.g., diadenosine tetraphosphate (Ap4A). Comprehensive reviews on P2Y receptor agonists and antagonists have recently appeared [7, 10, 49, 50]. Many of the nucleotides and their analogs, which have been developed as selective agonists for specific subtypes, are metabolically unstable under in vivo conditions. Therefore, it is advisable to study P2Y receptors using the frequently more stable, subtype-selective P2Y receptor antagonists, as far as available.

P2Y receptor subtypes
P2Y1 receptor antagonists
The nucleotide derivatives MRS2500 (29) and MRS2279 (30) are potent and selective antagonists that bind close to the orthosteric ADP binding site but have mostly allosteric interactions as shown in a co-crystal structure of 29 with the human P2Y1 receptor (see Table 2, Figs. 5 and 6) [51, 52]. In comparison to the antagonist 29, which bears only one phosphate group at the 5’-OH group and another one at the 3’-OH group of the adenosine analog (see Fig. 5), the agonist ADP and its derivative 2-methylthio-ADP (2MeSADP) are predicted to display a different binding mode, binding in a deeper orthosteric binding pocket [52]. This suggestion was based on molecular docking and molecular dynamics simulation (MD) studies taking into account an X-ray structure of the agonist-bound P2Y12 receptor, which is activated by the same agonists, ADP and 2MeSADP [52, 53]. The binding of the β-phosphate group of ADP to the basic amino acid residues R287, D304, and R310 is predicted to overlap with the binding of the 3’-phophate group of the antagonist MRS2500 (29) (see Fig. 6). The binding of an agonist is expected to contract the orthosteric binding pocket, thereby inducing an inward shift of the extracellular part of helix VI (stretching of the bent conformation) towards the center of the receptor. The antagonists 29 and 30, bearing phosphoric acid ester functions, are metabolically not very stable and are therefore not the first choice for in vivo studies but can be excellent tools for in vitro experiments.

Structures of selected P2Y receptor antagonists recommended as pharmacological tools or developed as drugs

Co-crystal structures of P2Y receptors and their ligands. For references see text
The urea derivative BPTU (31) is an allosteric antagonist that binds at the periphery of the receptor, far from the ADP binding site and close to the phospholipid interface (see Fig. 6) [52]. While the nucleotidic antagonists are highly water-soluble, 31 is lipophilic and displays only limited water solubility.
P2Y2 receptor antagonists
The most potent and selective P2Y2 receptor antagonist available so far is the nucleotide analog AR-C118925 (32) [54,55,56,57,58]. The competitive antagonist, whose structure imitates that of an uracil nucleotide, displays relatively high potency at human and rodent P2Y2 receptors and is well soluble in buffer at pH 7.4 due to its negatively charged tetrazolate ring which mimics a phosphate group [56]. Because of its high polarity, 32 has low peroral bioavailability and cannot penetrate into the brain. The compound showed high metabolic stability in human and mouse liver microsomes and is therefore suitable for in vivo studies but will have to be parenterally applied [52]. The P2Y2 receptor antagonist has already been used in a number of in vitro and in vivo studies to investigate the role of the receptor in health and disease (e.g., [59,60,61,62,63]).
P2Y4 receptor antagonists
Only few antagonists are available to study the P2Y4 receptor. PSB-16133 (33) and PSB-1699 (34) show P2Y4 receptor inhibition at submicromolar concentrations combined with selectivity versus the other P2Y receptor subtypes (see Fig. 5 and Table 2) [57]. PSB-16133 is somewhat more potent, while PSB-1699 is more selective for the P2Y4 receptor. Both compounds are sulfonated, negatively charged anthraquinone derivatives. These polar compounds display relatively good water solubility but can probably not penetrate cell membranes.
P2Y6 receptor antagonists
The isocyanato-substituted dimeric thiourea derivative MRS2578 (35) acts as an irreversible inhibitor of P2Y6 receptors [64]. The compound is not ideal due to its low water solubility and high reactivity, but nevertheless, it has already been utilized for a number of in vitro and in vivo studies (e.g., [65,66,67,68,69]).
P2Y11 receptor antagonists
Several suramin derivatives have been described to block the P2Y11 receptor with relatively high potency and selectivity [70]. One of the best antagonists so far is NF340 (36) (see Table 2 and Fig. 5), which acts as a competitive antagonist [70]. The compound bears four sulfonate functions and is therefore highly polar, deprotonated at pH 7.4, and well water-soluble in its deprotonated form.
P2Y12 receptor antagonists
The P2Y12 receptor is an important target for antithrombotic drugs, and P2Y12 antagonists are widely used to prevent cardiac infarction and stroke [71]. Therefore, a variety of P2Y12 receptor antagonists belonging to different chemical classes have been developed to date, and further drug discovery in this field is still ongoing.
The therapeutically used thienotetrahydropyridine derivatives clopidogrel and prasugrel are prodrugs of irreversibly acting allosteric P2Y12 receptor antagonists. They have to be metabolized by cytochrome P450 enzymes in the liver before they can react with a cysteine residue in the extracellular domain of the P2Y12 receptor that is not part of the orthosteric binding site, forming a stable disulfide bond [72, 73]. Therefore, these prodrugs are only suitable for systemic application in vivo where they can be metabolized in the liver. Only peripheral P2Y12 receptors will be blocked, preferably on thrombocytes.
For pharmacological studies of P2Y12 receptors, several direct, competitive antagonists are available, several of which are approved as therapeutic antithrombotic drugs endowed with the required pharmacokinetic properties. Cangrelor (37) is a therapeutically used nucleotide analog derived from ATP acting as a competitive antagonist and displaying very high potency and selectivity versus most P2Y receptor subtypes (see Fig. 5 and Table 2) [74]; however, it also potently blocks the P2Y13 receptor [75]. The compound is very polar and well water-soluble due to its nucleotidic structure; it can only be applied parenterally since it is not able to penetrate membranes and is therefore not orally bioavailable. Ticagrelor (38) is the result of cangrelor’s further optimization. It lacks the phosph(on)ate groups, is therefore less polar, and shows good peroral bioavailability. The compound is a potent competitive P2Y12 receptor antagonist with inverse agonistic activity that is widely employed as an antithrombotic drug. However, it was shown to additionally block the human adenosine A3 receptor and the equilibrative nucleoside transporter ENT-1 which might be responsible for side effects [76,77,78,79,80].
A different class of potent, competitive P2Y12 receptor antagonists bears a sulfoanthraquinone scaffold [81, 82]. PSB-0739 (39) was the most potent compound in this series with (sub)nanomolar potency and high selectivity. Due to its sulfonate groups, it is well water-soluble but not orally bioavailable. The compound has been applied in a number of experimental studies in vitro and in vivo to block P2Y12 receptors (e.g., [83,84,85,86]).
The acylsulfonamide derivative AZD 1283 (40) is another experimental drug that shows good water solubility [72, 87, 88]. The pKa value of its sulfonamide function is relatively low (4.6), and the compound is therefore deprotonated at pH 7.4. The competitive antagonist was co-crystallized with the human P2Y12 receptor (see Fig. 6) [72]. AZD 1283 binds in an elongated conformation making strong polar as well as hydrophobic interactions with the amino acid residues in the orthosteric binding pocket. Another sulfonamide derivative related to 40 is elinogrel (41) which was reported to also display high water solubility [89].
Selatogrel (ACT-246475, 42) belongs to a different class of competitive P2Y12 receptor antagonists with a peptide-like structure and a phosphonate function [89,90,91,92]. The drug itself is very polar and is therefore subcutaneously applied; an orally bioavailable prodrug, ACT-281959, has also been developed. Selatogrel is currently evaluated in clinical trials in patients with stable coronary artery disease and in patients with acute myocardial infarction.
Due to their polar nature, none of the currently available P2Y12 receptor antagonists penetrates well into the brain; if blocking of brain P2Y12 receptors is required, the drugs have to be directly injected. The development of a brain-permeant P2Y12 receptor antagonists would be highly desirable to study their potential for treating brain diseases, e.g., as neuroprotective drugs.
P2Y13 receptor antagonists
The P2Y13 receptor is the least investigated P2Y receptor subtype. It is most closely related to the P2Y12 receptor, and the P2Y12 receptor antagonist cangrelor (37), an ATP analog, was reported to also block the P2Y13 receptor subtype with high potency [75].
The pyridinecarboxaldehyde derivative MRS2211 (43), which bears a phosphate group, was described as a moderately potent competitive P2Y13 receptor antagonist, but its selectivity is unclear [93]. Since it bears an aldehyde function and a phosphoric acid ester group, its stability can be expected to be limited, and the compound is recommended for in vitro studies only.
P2Y14 receptor antagonists
Potent and selective competitive P2Y14 receptor antagonists have been developed [94, 95]. The commercially available antagonist PPTN (44), a naphthalenecarboxylic acid derivative, shows very high potency and selectivity [96].
Conclusions
The field of purinergic receptors, which was widely opened, nurtured, and continuously promoted by Geoffrey Burnstock, has now attracted researchers from many different areas of research. One of the latest disciplines that discovered the importance of purinergic signaling was immunology. And again, Burnstock saw its relevance long ago and paved the way [97]. It is now generally recognized that ATP and purinergic signaling are important for all processes in the body, in health, and especially in disease, where ATP can be described as a danger signal. P2Y12 receptor antagonists have become essential antithrombotic drugs. The first P2X3 receptor antagonist, gefapixant, named after Geoff Burnstock, is expected to be soon approved for the treatment of chronic cough. Other drugs interacting with P2 receptors are currently evaluated in clinical trials, and many more will be developed. The availability of suitable tool compounds and drugs will help to advance basic research and target validation in this field. While a number of tool compounds for studying P2X and P2Y receptors are available, they often lack drug-likeness, e.g., due to the presence of several negative charges associated with high polarity, large molecular weights, or moderate selectivity. Despite enormous progress in recent years, there is still much room for medicinal chemists to develop the ideal biological tool compounds and drugs for the large variety of P2 receptor subtypes.
Data availability
All data are available upon reasonable request.
Code availability
Not applicable.
References
Kennedy C (2021) The P2Y/P2X divide: how it began. Biochem Pharmacol 187:114408. https://doi.org/10.1016/j.bcp.2021.114408
Müller CE, Jacobson KA (2011) Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta 1808:1290–1308. https://doi.org/10.1016/j.bbamem.2010.12.017
Müller CE, Baqi Y, Namasivayam V (2020) Agonists and antagonists for purinergic receptors. Methods Mol Biol 2041:45–64. https://doi.org/10.1007/978-1-4939-9717-6_3
Thimm D, Schiedel AC, Peti-Peterdi J, Kishore BK, Müller CE (2015) The nucleobase adenine as a signalling molecule in the kidney. Acta Physiol (Oxf) 213:808–818. https://doi.org/10.1111/apha.12452
Zimmermann H (2021) Ectonucleoside triphosphate diphosphohydrolases and ecto-5’-nucleotidase in purinergic signaling: how the field developed and where we are now. Purinergic Signal 17:117–125. https://doi.org/10.1007/s11302-020-09755-6
Illes P, Müller CE, Jacobson KA, Grutter T, Nicke A, Fountain SJ, Kennedy C, Schmalzing G, Jarvis MF, Stojilkovic SS, King BF, Di Virgilio F (2021) Update of P2X receptor properties and their pharmacology: IUPHAR review 30. Br J Pharmacol 178:489–514. https://doi.org/10.1111/bph.15299
Jacobson KA, Müller CE (2016) Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 104:31–49. https://doi.org/10.1016/j.neuropharm.2015.12.001
Lambertucci C, Dal Ben D, Buccioni M, Marucci G, Thomas A, Volpini R (2015) Medicinal chemistry of P2X receptors: agonists and orthosteric antagonists. Curr Med Chem 22:915–928. https://doi.org/10.2174/0929867321666141215093513
Müller CE (2015) Medicinal chemistry of P2X receptors: allosteric modulators. Curr Med Chem 22:929–941. https://doi.org/10.2174/0929867322666141210155610
Jacobson KA, IJzerman AP, Müller CE (2021) Medicinal chemistry of P2 and adenosine receptors: common scaffolds adapted for multiple targets. Biochem Pharmacol 187:114311. https://doi.org/10.1016/j.bcp.2020.114311
Soto F, Lambrecht G, Nickel P, Stühmer W, Busch AE (1999) Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacology 38:141–149. https://doi.org/10.1016/s0028-3908(98)00158-0
Tian M, Abdelrahman A, Baqi Y, Fuentes E, Azazna D, Spanier C, Densborn S, Hinz S, Schmid R, Müller CE (2020) Discovery and structure relationships of salicylanilide derivatives as potent, non-acidic P2X1 receptor antagonists. J Med Chem 63:6164–6178. https://doi.org/10.1021/acs.jmedchem.0c00435
Wolf C, Rosefort C, Fallah G, Kassack MU, Hamacher A, Bodnar M, Wang H, Illes P, Kless A, Bahrenberg G, Schmalzing G, Hausmann R (2011) Molecular determinants of potent P2X2 antagonism identified by functional analysis, mutagenesis, and homology docking. Mol Pharmacol 79:649–661. https://doi.org/10.1124/mol.110.068700
Baqi Y, Hausmann R, Rosefort C, Rettinger J, Schmalzing G, Müller CE (2011) Discovery of potent competitive antagonists and positive modulators of the P2X2 receptor. J Med Chem 54:817–830. https://doi.org/10.1021/jm1012193
Schneider R, Leven P, Glowka T, Kuzmanov I, Lysson M, Schneiker B, Miesen A, Baqi Y, Spanier C, Grants I, Mazzotta E, Villalobos-Hernandez E, Kalff JC, Müller CE, Christofi FL, and Wehner S (2021) A novel P2X2-dependent purinergic mechanism of enteric gliosis in intestinal inflammation. EMBO Mol Med 13: e12724. https://doi.org/10.15252/emmm.202012724
Lee HJ, Joung SK, Kim YG, Yoo JY, Han SB (2004) Bioequivalence assessment of ambroxol tablet after a single oral dose administration to healthy male volunteers. Pharmacol Res 49:93–98. https://doi.org/10.1016/j.phrs.2003.07.011
Bouscary A, Quessada C, René F, Spedding M, Henriques A, Ngo S, Loeffler JP (2020) Drug repositioning in neurodegeneration: an overview of the use of ambroxol in neurodegenerative diseases. Eur J Pharmacol 884:173446. https://doi.org/10.1016/j.ejphar.2020.173446
Malerba M, Ragnoli B (2008) Ambroxol in the 21st century: pharmacological and clinical update. Expert Opin Drug Metab Toxicol 4:1119–1129. https://doi.org/10.1517/17425255.4.8.1119
Krajewski JL (2020) P2X3-containing receptors as targets for the treatment of chronic pain. Neurotherapeutics 17:826–838. https://doi.org/10.1007/s13311-020-00934-2
Ford AP (2012) In pursuit of P2X3 antagonists: novel therapeutics for chronic pain and afferent sensitization. Purinergic Signal 8:3–26. https://doi.org/10.1007/s11302-011-9271-6
Marucci G, Dal Ben D, Buccioni M, Martí Navia A, Spinaci A, Volpini R, Lambertucci C (2019) Update on novel purinergic P2X3 and P2X2/3 receptor antagonists and their potential therapeutic applications. Expert Opin Ther Pat 29:943–963. https://doi.org/10.1080/13543776.2019.1693542
Müller CE (2010) Emerging structures and ligands for P2X(3) and P2X(4) receptors-towards novel treatments of neuropathic pain. Purinergic Signal 6:145–148. https://doi.org/10.1007/s11302-010-9182-y
Morice AH, Birring SS, Smith JA, McGarvey LP, Schelfhout J, Martin Nguyen A, Xu ZJ, Wu WC, Muccino DR, Sher MR (2021) Characterization of patients with refractory or unexplained chronic cough participating in a phase 2 clinical trial of the P2X3-receptor antagonist gefapixant. Lung 199:121–129. https://doi.org/10.1007/s00408-021-00437-7
Dicpinigaitis PV (2021) Coming soon: the first-ever drug(s) for refractory chronic cough. Lung 199:83–84. https://doi.org/10.1007/s00408-021-00438-6
Morice A, Smith JA, McGarvey L, Birring SS, Parker SM, Turner A, Hummel T, Gashaw I, Fels L, Klein S, Francke K, and Friedrich C (2021) Eliapixant (BAY 1817080), a P2X3 receptor antagonist, in refractory chronic cough: a randomised, placebo-controlled, crossover phase 2a study. Eur Respir J. https://doi.org/10.1183/13993003.04240-2020
Burnstock G (2018) The therapeutic potential of purinergic signalling. Biochem Pharmacol 151:157–165. https://doi.org/10.1016/j.bcp.2017.07.016
Spinaci A, Buccioni M, Dal Ben D, Marucci G, Volpini R, Lambertucci C (2021) P2X3 receptor ligands: structural features and potential therapeutic applications. Front Pharmacol 12:653561. https://doi.org/10.3389/fphar.2021.653561
Wang J, Wang Y, Cui WW, Huang Y, Yang Y, Liu Y, Zhao WS, Cheng XY, Sun WS, Cao P, Zhu MX, Wang R, Hattori M, Yu Y (2018) Druggable negative allosteric site of P2X3 receptors. Proc Natl Acad Sci U S A 115:4939–4944. https://doi.org/10.1073/pnas.1800907115
Garceau D, Chauret N (2019) BLU-5937: a selective P2X3 antagonist with potent anti-tussive effect and no taste alteration. Pulm Pharmacol Ther 56:56–62. https://doi.org/10.1016/j.pupt.2019.03.007
Rommel C (2021) Paving the way for our future in science-based innovation. https://www.bayer.com/sites/default/files/BayerCMD2021_Pharma_RandD_Presentation.pdf. Accessed 8 July 2021
Friedrich C, Francke K, Birring SS, Van Den Berg JWK, Marsden P, Mcgarvey L, Turner A, Wielders P, Gashaw I, Klein S, Morice A (2020) Safety and efficacy of P2X3 antagonist BAY 1902607 in refractory chronic cough. Eur Respir J 56:4566. https://doi.org/10.1183/13993003.congress-2020.4566
Jarvis MF, Burgard EC, McGaraughty S, Honore P, Lynch K, Brennan TJ, Subieta A, van Biesen T, Cartmell J, Bianchi B, Niforatos W, Kage K, Yu H, Mikusa J, Wismer CT, Zhu CZ, Chu K, Lee C-H, Stewart AO, Polakowski J, Cox BF, Kowaluk E, Williams M, Sullivan J, Faltynek C (2002) A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci U S A 99:17179–17184. https://doi.org/10.1073/pnas.252537299
Mansoor SE, Lü W, Oosterheert W, Shekhar M, Tajkhorshid E, Gouaux E (2016) X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nature 538:66–71. https://doi.org/10.1038/nature19367
Abdelrahman A, Namasivayam V, Hinz S, Schiedel AC, Köse M, Burton M, El-Tayeb A, Gillard M, Bajorath J, de Ryck M, Müller CE (2017) Characterization of P2X4 receptor agonists and antagonists by calcium influx and radioligand binding studies. Biochem Pharmacol 125:41–54. https://doi.org/10.1016/j.bcp.2016.11.016
Matsumura Y, Yamashita T, Sasaki A, Nakata E, Kohno K, Masuda T, Tozaki-Saitoh H, Imai T, Kuraishi Y, Tsuda M, Inoue K (2016) A novel P2X4 receptor-selective antagonist produces anti-allodynic effect in a mouse model of herpetic pain. Sci Rep 6:32461. https://doi.org/10.1038/srep32461
Inoue K (2021) Nociceptive signaling of P2X receptors in chronic pain states. Purinergic Signal 17:41–47. https://doi.org/10.1007/s11302-020-09743-w
Teixeira JM, Dos Santos GG, Neves AF, Athie MCP, Bonet IJM, Nishijima CM, Farias FH, Figueiredo JG, Hernandez-Olmos V, Alshaibani S, Tambeli CH, Müller CE, Parada CA (2019) Diabetes-induced neuropathic mechanical hyperalgesia depends on P2X4 receptor activation in dorsal root ganglia. Neuroscience 398:158–170. https://doi.org/10.1016/j.neuroscience.2018.12.003
Hernandez-Olmos V, Abdelrahman A, El-Tayeb A, Freudendahl D, Weinhausen S, Müller CE (2012) N-substituted phenoxazine and acridone derivatives: structure-activity relationships of potent P2X4 receptor antagonists. J Med Chem 55:9576–9588. https://doi.org/10.1021/jm300845v
Ase AR, Honson NS, Zaghdane H, Pfeifer TA, Séguéla P (2015) Identification and characterization of a selective allosteric antagonist of human P2X4 receptor channels. Mol Pharmacol 87:606–616. https://doi.org/10.1124/mol.114.096222
Werner S, Mesch S, Hillig RC, Ter Laak A, Klint J, Neagoe I, Laux-Biehlmann A, Dahllöf H, Bräuer N, Puetter V, Nubbemeyer R, Schulz S, Bairlein M, Zollner TM, Steinmeyer A (2019) Discovery and characterization of the potent and selective P2X4 inhibitor N-[4-(3-Chlorophenoxy)-3-sulfamoylphenyl]-2-phenylacetamide (BAY-1797) and structure-guided amelioration of Its CYP3A4 induction profile. J Med Chem 62:11194–11217. https://doi.org/10.1021/acs.jmedchem.9b01304
Gelin CF, Bhattacharya A, Letavic MA (2020) P2X7 receptor antagonists for the treatment of systemic inflammatory disorders. Prog Med Chem 59:63–99. https://doi.org/10.1016/bs.pmch.2019.11.002
Rech JC, Bhattacharya A, Branstetter BJ, Love CJ, Leenaerts JE, Cooymans LP, Eckert WA 3rd, Ao H, Wang Q, Chaplan SR, Wickenden AD, Lebsack AD, Breitenbucher JG (2016) The discovery and preclinical characterization of 6-chloro-N-(2-(4,4-difluoropiperidin-1-yl)-2-(2-(trifluoromethyl)pyrimidin-5-yl)ethyl)quinoline-5-carboxamide based P2X7 antagonists. Bioorg Med Chem Lett 26:4781–4784. https://doi.org/10.1016/j.bmcl.2016.08.029
Karasawa A, and Kawate T (2016) Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 5: e22153. https://doi.org/10.7554/eLife.22153
Timmers M, Ravenstijn P, Xi L, Triana-Baltzer G, Furey M, Van Hemelryck S, Biewenga J, Ceusters M, Bhattacharya A, van den Boer M, van Nueten L, de Boer P (2018) Clinical pharmacokinetics, pharmacodynamics, safety, and tolerability of JNJ-54175446, a brain permeable P2X7 antagonist, in a randomised single-ascending dose study in healthy participants. J Psychopharmacol 32:1341–1350. https://doi.org/10.1177/0269881118800067
Hattori M, Gouaux E (2012) Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485:207–212. https://doi.org/10.1038/nature11010
Kawate T, Michel JC, Birdsong WT, Gouaux E (2009) Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature 460:592–598. https://doi.org/10.1038/nature08198
Kasuya G, Fujiwara Y, Tsukamoto H, Morinaga S, Ryu S, Touhara K, Ishitani R, Furutani Y, Hattori M, Nureki O (2017) Structural insights into the nucleotide base specificity of P2X receptors. Sci Rep 7:45208. https://doi.org/10.1038/srep45208
Jacobson KA, Delicado EG, Gachet C, Kennedy C, von Kügelgen I, Li B, Miras-Portugal MT, Novak I, Schöneberg T, Perez-Sen R, Thor D, Wu B, Yang Z, Müller CE (2020) Update of P2Y receptor pharmacology: IUPHAR Review 27. Br J Pharmacol 177:2413–2433. https://doi.org/10.1111/bph.15005
Rafehi M, Müller CE (2018) Tools and drugs for uracil nucleotide-activated P2Y receptors. Pharmacol Ther 190:24–80. https://doi.org/10.1016/j.pharmthera.2018.04.002
von Kügelgen I (2021) Molecular pharmacology of P2Y receptor subtypes. Biochem Pharmacol 187:114361. https://doi.org/10.1016/j.bcp.2020.114361
Kim HS, Ohno M, Xu B, Kim HO, Choi Y, Ji XD, Maddileti S, Marquez VE, Harden TK, Jacobson KA (2003) 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a northern conformation: enhanced potency as P2Y1 receptor antagonists. J Med Chem 46:4974–4987. https://doi.org/10.1021/jm030127+
Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, Han GW, Liu H, Cherezov V, Katritch V, Jiang H, Stevens RC, Jacobson KA, Zhao Q, Wu B (2015) Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520:317–321. https://doi.org/10.1038/nature14287
Li Y, Yin C, Liu P, Li D, Lin J (2017) Identification of a different agonist-binding site and activation mechanism of the human P2Y1 receptor. Sci Rep 7:13764. https://doi.org/10.1038/s41598-017-14268-1
Kemp PA, Sugar RA, Jackson AD (2004) Nucleotide-mediated mucin secretion from differentiated human bronchial epithelial cells. Am J Respir Cell Mol Biol 31:446–455. https://doi.org/10.1165/rcmb.2003-0211OC
Kindon N, Davis A, Dougall I, Dixon J, Johnson T, Walters I, Thom S, McKechnie K, Meghani P, Stocks MJ (2017) From UTP to AR-C118925, the discovery of a potent non nucleotide antagonist of the P2Y(2) receptor. Bioorg Med Chem Lett 27:4849–4853. https://doi.org/10.1016/j.bmcl.2017.09.043
Rafehi M, Burbiel JC, Attah IY, Abdelrahman A, Müller CE (2017) Synthesis, characterization, and in vitro evaluation of the selective P2Y(2) receptor antagonist AR-C118925. Purinergic Signal 13:89–103. https://doi.org/10.1007/s11302-016-9542-3
Rafehi M, Malik EM, Neumann A, Abdelrahman A, Hanck T, Namasivayam V, Müller CE, Baqi Y (2017) Development of potent and selective antagonists for the UTP-activated P2Y(4) receptor. J Med Chem 60:3020–3038. https://doi.org/10.1021/acs.jmedchem.7b00030
Rafehi M, Neumann A, Baqi Y, Malik EM, Wiese M, Namasivayam V, Müller CE (2017) Molecular recognition of agonists and antagonists by the nucleotide-activated G protein-coupled P2Y(2) receptor. J Med Chem 60:8425–8440. https://doi.org/10.1021/acs.jmedchem.7b00854
Zhang Y, Ecelbarger CM, Lesniewski LA, Müller CE, Kishore BK (2020) P2Y(2) receptor promotes high-fat diet-induced obesity. Front Endocrinol (Lausanne) 11:341. https://doi.org/10.3389/fendo.2020.00341
Woods LT, Jasmer KJ, Muñoz Forti K, Shanbhag VC, Camden JM, Erb L, Petris MJ, Weisman GA (2020) P2Y(2) receptors mediate nucleotide-induced EGFR phosphorylation and stimulate proliferation and tumorigenesis of head and neck squamous cell carcinoma cell lines. Oral Oncol 109:104808. https://doi.org/10.1016/j.oraloncology.2020.104808
Magni G, Merli D, Verderio C, Abbracchio MP, Ceruti S (2015) P2Y2 receptor antagonists as anti-allodynic agents in acute and sub-chronic trigeminal sensitization: role of satellite glial cells. Glia 63:1256–1269. https://doi.org/10.1002/glia.22819
Hochhauser E, Cohen R, Waldman M, Maksin A, Isak A, Aravot D, Jayasekara PS, Müller CE, Jacobson KA, Shainberg A (2013) P2Y2 receptor agonist with enhanced stability protects the heart from ischemic damage in vitro and in vivo. Purinergic Signal 9:633–642. https://doi.org/10.1007/s11302-013-9374-3
Wang S, Iring A, Strilic B, Albarrán Juárez J, Kaur H, Troidl K, Tonack S, Burbiel JC, Müller CE, Fleming I, Lundberg JO, Wettschureck N, Offermanns S (2015) P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J Clin Invest 125:3077–3086. https://doi.org/10.1172/jci81067
Mamedova LK, Joshi BV, Gao ZG, von Kügelgen I, Jacobson KA (2004) Diisothiocyanate derivatives as potent, insurmountable antagonists of P2Y6 nucleotide receptors. Biochem Pharmacol 67:1763–1770. https://doi.org/10.1016/j.bcp.2004.01.011
Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K (2007) UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446:1091–1095. https://doi.org/10.1038/nature05704
Wen RX, Shen H, Huang SX, Wang LP, Li ZW, Peng P, Mamtilahun M, Tang YH, Shen FX, Tian HL, Yang GY, Zhang ZJ (2020) P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci Ther 26:416–429. https://doi.org/10.1111/cns.13296
Oliveira-Giacomelli Á, Albino CM, de Souza HDN, Corrêa-Velloso J, de Jesus Santos AP, Baranova J, Ulrich H (2019) P2Y6 and P2X7 receptor antagonism exerts neuroprotective/neuroregenerative effects in an animal model of parkinson’s disease. Front Cell Neurosci 13:476. https://doi.org/10.3389/fncel.2019.00476
Huang D, Yang J, Liu X, He L, Luo X, Tian H, Xu T, Zeng J (2018) P2Y(6) receptor activation is involved in the development of neuropathic pain induced by chronic constriction injury of the sciatic nerve in rats. J Clin Neurosci 56:156–162. https://doi.org/10.1016/j.jocn.2018.07.013
Müller T, Fay S, Vieira RP, Karmouty-Quintana H, Cicko S, Ayata CK, Zissel G, Goldmann T, Lungarella G, Ferrari D, Di Virgilio F, Robaye B, Boeynaems JM, Lazarowski ER, Blackburn MR, Idzko M (2017) P2Y(6) receptor activation promotes inflammation and tissue remodeling in pulmonary fibrosis. Front Immunol 8:1028. https://doi.org/10.3389/fimmu.2017.01028
Meis S, Hamacher A, Hongwiset D, Marzian C, Wiese M, Eckstein N, Royer HD, Communi D, Boeynaems JM, Hausmann R, Schmalzing G, Kassack MU (2010) NF546 [4,4’-(carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)-carbonylimino))-bis(1,3-xylene-alpha, alpha’-diphosphonic acid) tetrasodium salt] is a non-nucleotide P2Y11 agonist and stimulates release of interleukin-8 from human monocyte-derived dendritic cells. J Pharmacol Exp Ther 332:238–247. https://doi.org/10.1124/jpet.109.157750
Baqi Y, Müller CE (2019) Antithrombotic P2Y(12) receptor antagonists: recent developments in drug discovery. Drug Discov Today 24:325–333. https://doi.org/10.1016/j.drudis.2018.09.021
Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q (2014) Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 509:115–118. https://doi.org/10.1038/nature13083
Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP, Pascal M, Herbert JM (2000) Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost 84:891–896
Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W (1999) Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem 42:213–220. https://doi.org/10.1021/jm981072s
Marteau F, Le Poul E, Communi D, Communi D, Labouret C, Savi P, Boeynaems JM, Gonzalez NS (2003) Pharmacological characterization of the human P2Y13 receptor. Mol Pharmacol 64:104–112. https://doi.org/10.1124/mol.64.1.104
Springthorpe B, Bailey A, Barton P, Birkinshaw TN, Bonnert RV, Brown RC, Chapman D, Dixon J, Guile SD, Humphries RG, Hunt SF, Ince F, Ingall AH, Kirk IP, Leeson PD, Leff P, Lewis RJ, Martin BP, McGinnity DF, Mortimore MP, Paine SW, Pairaudeau G, Patel A, Rigby AJ, Riley RJ, Teobald BJ, Tomlinson W, Webborn PJ, Willis PA (2007) From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett 17:6013–6018. https://doi.org/10.1016/j.bmcl.2007.07.057
James S, Akerblom A, Cannon CP, Emanuelsson H, Husted S, Katus H, Skene A, Steg PG, Storey RF, Harrington R, Becker R, Wallentin L (2009) Comparison of ticagrelor, the first reversible oral P2Y(12) receptor antagonist, with clopidogrel in patients with acute coronary syndromes: rationale, design, and baseline characteristics of the PLATelet inhibition and patient Outcomes (PLATO) trial. Am Heart J 157:599–605. https://doi.org/10.1016/j.ahj.2009.01.003
Hoffmann K, Lutz DA, Straßburger J, Baqi Y, Müller CE, von Kügelgen I (2014) Competitive mode and site of interaction of ticagrelor at the human platelet P2Y12-receptor. J Thromb Haemost 12:1898–1905. https://doi.org/10.1111/jth.12719
Olivier CB, Diehl P, Schnabel K, Weik P, Zhou Q, Bode C, Moser M (2014) Third generation P2Y12 antagonists inhibit platelet aggregation more effectively than clopidogrel in a myocardial infarction registry. Thromb Haemost 111:266–272. https://doi.org/10.1160/th13-06-0508
Armstrong D, Summers C, Ewart L, Nylander S, Sidaway JE, van Giezen JJ (2014) Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J Cardiovasc Pharmacol Ther 19:209–219. https://doi.org/10.1177/1074248413511693
Baqi Y, Atzler K, Köse M, Glänzel M, Müller CE (2009) High-affinity, non-nucleotide-derived competitive antagonists of platelet P2Y12 receptors. J Med Chem 52:3784–3793. https://doi.org/10.1021/jm9003297
Hoffmann K, Baqi Y, Morena MS, Glänzel M, Müller CE, von Kügelgen I (2009) Interaction of new, very potent non-nucleotide antagonists with arg256 of the human platelet P2Y12 receptor. J Pharmacol Exp Ther 331:648–655. https://doi.org/10.1124/jpet.109.156687
Micklewright JJ, Layhadi JA, Fountain SJ (2018) P2Y(12) receptor modulation of ADP-evoked intracellular Ca(2+) signalling in THP-1 human monocytic cells. Br J Pharmacol 175:2483–2491. https://doi.org/10.1111/bph.14218
de Almeida-Pereira L, Repossi MG, Magalhães CF, Azevedo RF, Corrêa-Velloso JDC, Ulrich H, Ventura ALM, Fragel-Madeira L (2018) P2Y(12) but not P2Y(13) purinergic receptor controls postnatal rat retinogenesis in vivo. Mol Neurobiol 55:8612–8624. https://doi.org/10.1007/s12035-018-1012-1
Bekő K, Koványi B, Gölöncsér F, Horváth G, Dénes Á, Környei Z, Botz B, Helyes Z, Müller CE, Sperlágh B (2017) Contribution of platelet P2Y(12) receptors to chronic Complete Freund’s adjuvant-induced inflammatory pain. J Thromb Haemost 15:1223–1235. https://doi.org/10.1111/jth.13684
Kishore BK, Carlson NG, Ecelbarger CM, Kohan DE, Müller CE, Nelson RD, Peti-Peterdi J, Zhang Y (2015) Targeting renal purinergic signalling for the treatment of lithium-induced nephrogenic diabetes insipidus. Acta Physiol (Oxf) 214:176–188. https://doi.org/10.1111/apha.12507
Bach P, Antonsson T, Bylund R, Björkman JA, Österlund K, Giordanetto F, van Giezen JJ, Andersen SM, Zachrisson H, Zetterberg F (2013) Lead optimization of ethyl 6-aminonicotinate acyl sulfonamides as antagonists of the P2Y12 receptor. separation of the antithrombotic effect and bleeding for candidate drug AZD1283. J Med Chem 56:7015–7024. https://doi.org/10.1021/jm400820m
Zhou S, Fang D, Tan S, Lin W, Wu W, Zheng K (2017) Investigating the binding mechanism of novel 6-aminonicotinate-based antagonists with P2Y(12) by 3D-QSAR, docking and molecular dynamics simulations. J Biomol Struct Dyn 35:2938–2965. https://doi.org/10.1080/07391102.2016.1237381
Zech G, Hessler G, Evers A, Weiss T, Florian P, Just M, Czech J, Czechtizky W, Görlitzer J, Ruf S, Kohlmann M, Nazaré M (2012) Identification of high-affinity P2Y12 antagonists based on a phenylpyrazole glutamic acid piperazine backbone. J Med Chem 55:8615–8629. https://doi.org/10.1021/jm300771j
Parlow JJ, Burney MW, Case BL, Girard TJ, Hall KA, Harris PK, Hiebsch RR, Huff RM, Lachance RM, Mischke DA, Rapp SR, Woerndle RS, Ennis MD (2010) Piperazinyl glutamate pyridines as potent orally bioavailable P2Y12 antagonists for inhibition of platelet aggregation. J Med Chem 53:2010–2037. https://doi.org/10.1021/jm901518t
Crescence L, Darbousset R, Caroff E, Hubler F, Riederer MA, Panicot-Dubois L, Dubois C (2021) Selatogrel, a reversible P2Y12 receptor antagonist, has reduced off-target interference with haemostatic factors in a mouse thrombosis model. Thromb Res 200:133–140. https://doi.org/10.1016/j.thromres.2021.01.026
Milluzzo RP, Franchina GA, Capodanno D, Angiolillo DJ (2020) Selatogrel, a novel P2Y(12) inhibitor: a review of the pharmacology and clinical development. Expert Opin Investig Drugs 29:537–546. https://doi.org/10.1080/13543784.2020.1764533
Kim YC, Lee JS, Sak K, Marteau F, Mamedova L, Boeynaems JM, Jacobson KA (2005) Synthesis of pyridoxal phosphate derivatives with antagonist activity at the P2Y13 receptor. Biochem Pharmacol 70:266–274. https://doi.org/10.1016/j.bcp.2005.04.021
Barrett MO, Sesma JI, Ball CB, Jayasekara PS, Jacobson KA, Lazarowski ER, Harden TK (2013) A selective high-affinity antagonist of the P2Y14 receptor inhibits UDP-glucose-stimulated chemotaxis of human neutrophils. Mol Pharmacol 84:41–49. https://doi.org/10.1124/mol.113.085654
Jung YH, Salmaso V, Wen Z, Bennett JM, Phung NB, Lieberman DI, Gopinatth V, Randle JCR, Chen Z, Salvemini D, Karcz TP, Cook DN, Jacobson KA (2021) Structure-activity relationship of heterocyclic P2Y(14) receptor antagonists: removal of the zwitterionic character with piperidine bioisosteres. J Med Chem 64:5099–5122. https://doi.org/10.1021/acs.jmedchem.1c00164
Gauthier JY, Belley M, Deschênes D, Fournier JF, Gagné S, Gareau Y, Hamel M, Hénault M, Hyjazie H, Kargman S, Lavallée G, Levesque JF, Li L, Mamane Y, Mancini J, Morin N, Mulrooney E, Robichaud J, Thérien M, Tranmer G, Wang Z, Wu J, Black WC (2011) The identification of 4,7-disubstituted naphthoic acid derivatives as UDP-competitive antagonists of P2Y14. Bioorg Med Chem Lett 21:2836–2839. https://doi.org/10.1016/j.bmcl.2011.03.081
Burnstock G, Boeynaems JM (2014) Purinergic signalling and immune cells. Purinergic Signal 10:529–564. https://doi.org/10.1007/s11302-014-9427-2
Funding
Open Access funding enabled and organized by Projekt DEAL. Part of our studies on P2X receptors was funded by the German Federal Ministry of Education and Research (BMBF). The authors are supported by the Deutsche Forschungsgemeinschaft through the Collaborative Research Center — SFB 1328 “Adenine Nucleotides in Immunity and Inflammation.”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
We hereby confirm that we have complied with ethical standards.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is dedicated to the memory of Professor Dr. Geoffrey Burnstock, a brilliant, free-spirited, creative scientist, a gifted artist, a dedicated teacher, a true cosmopolitan, a connoisseur of human nature and passionate philanthrope, and an irreplaceable colleague and friend.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Müller, C.E., Namasivayam, V. Recommended tool compounds and drugs for blocking P2X and P2Y receptors. Purinergic Signalling 17, 633–648 (2021). https://doi.org/10.1007/s11302-021-09813-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-021-09813-7