
Abstract
The selective, high affinity A2B adenosine receptor (AdoR) antagonists that were synthesized by several research groups should aid in determining the role of the A2B AdoR in inflammatory diseases like asthma or rheumatoid arthritis (RA) and angiogenic diseases like diabetic retinopathy or cancer. CV Therapeutics scientists discovered the selective, high affinity A2B AdoR antagonist 10, a 8-(4-pyrazolyl)-xanthine derivative [CVT-6883, Ki(hA2B) = 22 nM; Ki(hA1) = 1,940 nM; Ki(hA2A) = 3,280; and Ki(hA3) = 1,070 nM] that has favorable pharmacokinetic (PK) properties (t 1/2 = 4 h and F > 35% rat). Compound 10 demonstrated functional antagonism at the A2B AdoR (KB = 6 nM) and efficacy in a mouse model of asthma. In two phase 1 clinical trials, CVT-6883 was found to be safe, well tolerated, and suitable for once daily dosing. A second compound 20, 8-(5-pyrazolyl)-xanthine, has been nominated for development from Baraldi’s group in conjunction with King Pharmaceuticals that has favorable A2B AdoR affinity and selectivity [Ki(hA2B) = 5.5 nM; Ki(hA1) > 1,000 nM; Ki(hA2A) > 1,000; and Ki(hA3) > 1,000 nM], and it has been demonstrated to be a functional antagonist. A third compound 32, a 2-aminopyrimidine, from the Almirall group has high A2B AdoR affinity and selectivity [Ki(hA2B) = 17 nM; Ki(hA1) > 1,000 nM; Ki(hA2A) > 2,500; and Ki(hA3) > 1,000 nM], and 32 has been moved into preclinical safety testing. Since three highly selective, high affinity A2B AdoR antagonists have been nominated for development with 10 (CVT-6883) being the furthest along in the development process, the role of the A2B AdoR in various disease states will soon be established.
Similar content being viewed by others

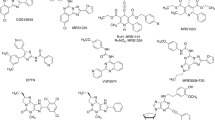
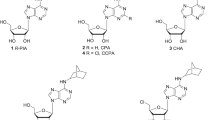
Avoid common mistakes on your manuscript.
Introduction
The need for a selective, high affinity A2B adenosine receptor (AdoR) antagonist, to fully establish the therapeutic potential of this class of agents as anti-inflammatory and antiangiogenic agents, has attracted the interest of several medicinal chemistry groups around the world [1–11]. The structural approach taken by these groups can be divided into two classes of compounds, xanthines and non-xanthine derivatives. The xanthine derivatives caffeine and theophylline are considered classic nonselective antagonists for adenosine receptors (Fig. 1). Theophylline 1, which has 9 µM affinity for the A2B AdoR, displays no selectivity against the other AdoRs [12]. Enprofylline 2, a 3-propyl xanthine derivative has moderate A2B affinity and low selectivity over the other AdoRs. Following further structural exploration of the xanthine moiety by several groups, the discovery of 8-phenylxanthines as selective A2B AdoR antagonists was made [13–15]. Among these 8-phenylxanthine derivatives, p-cyanoanilide 3 (MRS-1754) of Jacobson et al. [16] and a negatively charged compound 4 (PSB-1115) of Muller et al. [17] stand out as selective A2B AdoR antagonists. To address the metabolic stability of compound 3 in human liver microsomal enzymes, Zablocki et al. [18] synthesized compound 5 (CVT-5440) that contains a bioisostere of the metabolically labile amide group present in 3. Compound 5 demonstrated good affinity for the A2B AdoR and selectivity over the other AdoRs. Improved in vitro metabolic stability was also observed in 5 compared to 3, but 5 still has a very low systemic exposure in rats when dosed orally, presumably due to low solubility.

Classic and prototypical xanthine-derived A2B receptor antagonists
Xanthines
CV Therapeutics (CVT) chemists started with these initial leads in their search for the discovery of a selective, high affinity A2B AdoR antagonist with good pharmaceutical properties [19, 20]. Kalla et al. [21] have explored various heterocycles as bioisosteric replacements for the phenyl group at the 8-position of xanthine and discovered that the 8-(pyrazol-4-yl)xanthines display good A2B AdoR affinity (Fig. 2). The prototypical compound 1,3-dipropyl-8-(1H-pyrazol-4-yl)xanthine 6 (CVT-5450) has high A2B AdoR affinity (9 nM), but displayed very low selectivity. Following oral dosing in rats, 6 displayed very high levels of systemic exposure; this encouraged CVT chemists to probe the 8-(pyrazol-4-yl)xanthine ligand to increase the selectivity [8]. Benzyl substitution on the pyrazole ring increased the selectivity compared to the phenyl, phenethyl, and phenpropyl derivatives. Optimization of the phenyl ring substitution suggested that the electron withdrawing groups F and CF3 at the meta-position increased selectivity toward the A2B AdoR. Compound 7, 1,3-dipropyl-8-(1-(3-(trifluoromethyl)benzyl)-1H-pyrazol-4-yl)xanthine that has 3-CF3 benzyl substitution on the pyrazole ring, displayed better selectivity compared to the unsubstituted derivative 6. Replacing the 1,3-dipropyl groups of the xanthine core with various alkyl groups like methyl, ethyl, butyl, and isobutyl groups suggested that smaller alkyl groups relative to propyl increase the A2B AdoR affinity and selectivity compared to the large groups. Compound 1,3-dimethyl-8-(1-(3-(trifluoromethyl)benzyl)-1H-pyrazol-4-yl)-xanthine 8 (CVT-6975) has very high A2B AdoR affinity and selectivity [21]. This observation prompted further investigation of the differential alkyl substitution at N-1 and N-3 positions [22]. Compound 9 displayed better affinity and selectivity compared to the dipropyl derivative 7, but has weaker affinity and selectivity compared to the dimethyl derivative 8. The 3-ethyl-1-propyl-8-(1-(3-(trifluoromethyl)benzyl)-1H-pyrazol-4-yl)-xanthine 10 (CVT-6883) has very good A2B AdoR affinity, and also it displayed good selectivity over other AdoR subtypes [22].

CVT A2B adenosine receptor antagonists—8-(pyrazol-4-yl) xanthines
Investigation of the monosubstitution at the N-1 position of the 8-pyrazolyl xanthine delivered a very high affinity and selective A2B AdoR antagonists [23]. For example, the 1-propyl-8-(1-(3-(trifluoromethyl)benzyl)-1H-pyrazol-4-yl)-xanthine 11 (CVT-7124) displays high A2B AdoR affinity (6 nM) and very good selectivity. This further supports the Hayallah et al. observation in the 8-phenyl xanthine series of compounds, that the monosubstitution at the N-1 position of the xanthine core enhances the A2B AdoR selectivity [17].
Elzein et al. replaced the phenyl group of 7 with different heterocycles including 3-phenyl-1,2,4-oxadiazoles, 5-phenyl-1,2,4-oxadiazoles and 3-phenyl-isoxazoles as these groups in the 8-phenyl xanthine series [18] improved the selectivity for the A2B AdoR receptor (Fig. 3) [24]. In this series, all the compounds display very good selectivity regardless of the substitutions at the N-1 and N-3 positions of the xanthine core. The 1,3-dipropyl analogue 8-(1-((5-(4-chlorophenyl)-1,2,4-oxadiazol-3-yl)methyl)-1H-pyrazol-4-yl)-xanthine 12 and N-1 propyl, N-3 ethyl analogues 3-ethyl-1-propyl-8-(1-((5-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)methyl)-xanthine 13 and 3-ethyl-1-propyl-8-(1-((5-(4-(trifluoromethyl)phenyl) isoxazol-3-yl)methyl)-1H-pyrazol-4-yl)-xanthine 14 display high affinity and selectivity for the A2B AdoR. Similar to the phenyl series of compounds, the N-1 monosubstituted oxadiazole and isoxazole derivatives of 8-pyrazolyl xanthines displayed high affinity and selectivity for the A2B AdoR. The N-1 propyl derivative 15 (CVT-6694) has a very high A2B affinity (7 nM) and very weak affinity for the A1, A2A, and A3 AdoRs [23]. The cyclopropyl methyl analogues 16 and 17 also displayed high affinity and selectivity for the A2B AdoR.

CVT A2B adenosine receptor antagonists—8-(pyrazol-4-yl)xanthines
In summary, CVT chemists discovered several high affinity and selective A2B AdoR antagonists. The pharmacophore, 8-(pyrazol-4-yl)xanthine, identified by the CVT chemists can provide selective A2B AdoR antagonists depending on the substitution pattern. From the above compounds, two selective antagonists 10 (CVT-6883) and 15 (CVT-6694) were chosen for further evaluation of the pharmacological and pharmaceutical properties. Compound 10 antagonized the 5′-N-ethylcarboxamidoadenosine (NECA)-induced cyclic adenosine monophosphate (cAMP) accumulation in human embryonic kidney (HEK)-A2B cells and NIH 3T3 cells, and compound 15 completely abolished the NECA-induced cAMP accumulation in bronchial smooth muscle cells (BSMCs) [25] proving that these compounds are functioning as antagonists for the hA2B AdoR. Compound 10, when dosed orally in rats at 2 mg/kg, displayed excellent systemic exposure with a Cmax 1,100 ng/ml and dose-adjusted area under curve (dAUC) 6,500 ng.h/ml [22] with a long half-life of 4 h (IV dosing, rat). When dosed orally in rats compound 15 exhibited very low systemic exposure. Therefore, compound 10 was selected as a lead molecule and moved into CVT’s development program.
Baraldi’s group evaluated a series of 8-heterocyclic substituted xanthines as antagonists for the A2B AdoR [26]. Of these derivatives, 8-(pyrazol-5-yl)xanthine derivatives displayed high affinity and selectivity for the A2B AdoR (Fig. 4). These 5-pyrazolyl derivatives 18 and 19 showed good affinity for the A2B AdoR and selectivity over other AdoR subtypes [27]. Both compounds block NECA-induced cAMP accumulation with IC50 values in the nanomolar range. Further exploration of the 5-pyrazolyl class resulted in a lead compound 20 (MRE-2029-F20) that has high affinity and selectivity for the A2B AdoR. The tritium-labeled derivative 21 ([3H]MRE-2029-F20) displayed a K D value of 1.65 ± 0.10 nM in Chinese hamster ovary (CHO) cells expressing hA2B receptors, and it can be useful as a pharmacological tool in binding studies [28].

Baraldi’s A2B adenosine receptor antagonists
In recent patent applications, Adenosine Therapeutics described a series of 8-pyridyl substituted xanthines as A2B AdoR antagonists (Fig. 5) [29]. The 8-pyridyl was further extended by substitution with heteroaryl (23 and 25), heterocyclyl (22), or alaninol (24) groups. According to the patent applications, some of these derivatives (22–25) have an A2B AdoR affinity of < 100 nM, but no selectivity data were given, so it is hard to completely evaluate the series.

Adenosine Therapeutics A2B adenosine receptor antagonists
9-Deazaxanthines
9-Deazaxanthines (pyrrolo[2,3-d]pyrimidinones) were initially explored by Grahner et al. as antagonists for the A1 and A2 AdoRs (Fig. 6) [30]. In most cases, the authors observed that the structure-activity relationships (SAR) of 9-deazaxanthines are parallel to those of xanthine derivatives and also noticed an increased selectivity over A1 AdoR. The authors concluded that the xanthines and 9-deazaxanthines bind in the same mode to the adenosine receptors, and thus, the similar SAR. Hayallah et al. have investigated the N-1 monosubstituted 9-deazaxanthines, because the corresponding xanthines generally exhibit high A2B AdoR selectivity [17]. The compound 6-phenyl-3-propyl-1H-pyrrolo[3,2-d]pyrimidine-2,4(3H,5H)-dione 26 has displayed good A2B AdoR affinity, but it did not exhibit good selectivity over the A1 AdoR as expected. Vidal et al. have synthesized 8-phenyl-9-deazaxanthines that have a sulfonamide linker at the para-position of the phenyl group, and many compounds exhibited good A2B AdoR affinity [31]. For instance, 27 of the above series displayed 6 nM affinity for the A2B AdoR and displayed good selectivity. In a recent publication, Carotti et al. presented several 9-deazaxanthines that have piperidine amides and piperazine amide substitution at the para-position of the 8-phenyl group [32]. Representatives from these classes, compounds 28 and 29 (Fig. 6), respectively, displayed both high affinity and selectivity for the A2B AdoR. CVT chemists have explored the 8-pyrazolyl-9-deazaxanthines as A2B AdoR antagonists [33]. The m-F benzyl derivative 30, 6-(1-(3-fluorobenzyl)-1H-pyrazol-4-yl)-1,3-dipropyl-1H-pyrrolo[3,2-d]pyrimidine-2,4(3H,5H)-dione, has good A2B AdoR affinity, but it did not offer good selectivity over other the AdoR subtypes. The corresponding m-CF3 benzyl derivative 31 displayed lower affinity for the A2B receptor than 30, but it exhibited good selectivity for the A2B AdoR. Overall the 9-deazaxanthines afforded similar SAR to the parent xanthines with respect to A2B AdoR affinity and, in most cases, higher selectivity.

Deazaxanthines as A2B antagonists
Non-xanthine analogues
Two series of compounds, 2-aminopyridines and 2-aminopyrimidines, were published as A2B AdoR antagonists in patent applications from Almirall Prodesfarma (Fig. 7) [34, 35]. From these series of compounds, Vidal et al. recently published on the common core and substituents, namely, N-heteroaryl 4′-furyl-4,5′-bipyrimidin-2′-amines, as high affinity and selective A2B AdoR antagonists [36]. For example, the 2′-amino(3-pyridyl) derivative 32 (LAS38096) has a A2B affinity of 17 nM and has very good selectivity. Similar analogues, 2′-amino(5-pyrimidinyl) derivative 33 and 2′-amino(6-oxo-1,6-dihydropyridin-3-yl) derivative 34, displayed good A2B affinity of 24 and 16 nM, respectively, and both compounds have very good A2B AdoR selectivity as well. Compound 32 inhibited the NECA-induced cAMP levels in HEK-293 expressing human A2B AdoR and CHO cells transfected with mouse A2B AdoR with IC50s of 321 nM and 349 nM, respectively. Following oral dosing in rats (10 mg/kg), compound 32 displayed good systemic exposure with a Cmax of 11 µM and an AUC of 16 µM/h. It also displayed good exposure following oral dosing in mouse and dogs. Based on its in vitro pharmacology and pharmacokinetic profile, 32 was moved into preclinical development.

Almirall A2B antagonists
Adenine derivatives have been explored as adenosine receptor antagonists by several research groups (Fig. 8) [37, 38]. Cristalli and coworkers reported a series of 2-substituted 9-alkyl derivatives as selective A2B receptor antagonists (not shown) [39]. Harada et al. at Eisai explored the 2-alkynyl-8-aryl-9-methyl adenine derivatives as A2B AdoR antagonists [40]. Of these derivatives, compound 35 with a 3-F phenyl substitution at the 8-position displayed good A2B affinity, but no binding selectivity over other AdoR subtypes (Fig. 8). Substituting the 3-F phenyl of 35 with a 2-furyl group provided compound 36 with good A2B affinity, but again with no selectivity. Further optimization of the 9-position of the adenine derivative 35 led to the 3-benzamide derivative 37 with excellent A2B affinity [41]. Compound 37 displayed good selectivity over the A1 AdoR subtype only. These analogues inhibited NECA-induced cAMP production in CHO K1 cells expressing the human A2B AdoR demonstrating that these compounds are antagonists. Further optimization of the SAR may lead to selective A2B antagonists in the adenine series.

Purines and 7-deazapurines as A2B antagonists
In recent publications, scientists at OSI Pharmaceuticals have shown that 2-phenyl-7-deazaadenines (pyrrolopyrimidines) display good A2B AdoR affinity (Fig. 8) [42]. A lead compound 38 in the pyrrolopyrimidine series demonstrated excellent A2B AdoR affinity and promising selectivity. A tritium-labeled analogue 39 ([3H]OSIP-339391) of 38 was synthesized, which displayed a K D value of 0.41 ± 0.06 nM for binding to human A2B AdoR expressed in HEK-293 cells. This represents a selective and high affinity radioligand that can be a useful tool in further characterization of the pharmacology of the A2B AdoR.
Pharmacology discussion
Since the goal of obtaining a high affinity and selective A2B antagonist has been achieved by several research groups, the agents obtained have been used to establish the anti-inflammatory properties in both in vitro cellular studies and in asthma models. CVT chemists have synthesized several A2B-selective antagonists including 15 (CVT-6694) and 10 (CVT-6883). Following stimulation with a nonselective agonist NECA, compound 15 attenuated the increased production of both interleukin (IL)-6 and monocyte chemotactic protein-1 (MCP-1) in bronchoalveolar lavage smooth muscle cells [25]. These experiments suggest a novel mechanism whereby adenosine acts as a proinflammatory mediator in the bronchiole airways. Similarly, A2B AdoR subtype is the predominant AdoR expressed in human lung fibroblasts (HLFs), which on activation by NECA increases the release of IL-6 in a concentration-dependent manner and induces the differentiation of fibroblast into myofibroblasts [43]. Synergy exists between hypoxia and NECA activation of the A2B AdoR in HLFs, thus resulting in a pronounced increase in the release of IL-6. The A2B antagonist 15 completely abolished the augmented effect of NECA on the IL-6 release; however, it as expected did not affect the hypoxia-induced release of IL-6 [44]. In a mouse asthma model (ragweed challenge), compound 10 (dose: 1 mg/kg IP, 14-day treatment) was as effective as montelukast in reducing AMP-induced airway reactivity [48]. Compound 10 reduced significantly bleomycin (3.0 U/kg)-induced pulmonary fibrosis and inflammation in mice [47]. Furthermore, 10 (dose: 1 mg/kg IP b.i.d.) relative to vehicle controls reduced lung fibrosis and levels of macrophage-derived mediators of lung remodeling [IL-6, osteopontin (OPN), transforming growth factor (TGF)-β1, and matrix metalloproteases (MMPs)] in adenosine deaminase-deficient (ADA -/-) mice [47].
The selective A2B AdoR antagonist, MRE-2029-F20 synthesized by Baraldi’s group, shows the inhibition of cAMP levels in neutrophils, lymphocytes, and HMC1 cells that naturally express the A2B AdoR that may play a role in inflammatory diseases [45]. The selective A2B AdoR antagonist 32 (LAS38096) synthesized by Almirall has been shown to inhibit the NECA-induced production of IL-6 in a dose-dependent manner in both human and mouse fibroblasts [36]. This further confirms the anti-inflammatory properties of A2B AdoR antagonists.
Conclusion
Compound 10 (CVT-6883), a potent selective, orally available, and potentially first in class A2B AdoR antagonist, has been entered into clinical trials by CV Therapeutics [46]. The data from two phase 1 clinical trials, a single ascending dose study in 24 healthy volunteers and a multiple ascending dose study in 30 volunteers, demonstrated that CVT-6883 was safe and well tolerated with no serious adverse events reported. Furthermore, the pharmacokinetic results indicated the suitability of CVT-6883 for once daily chronic dosing. The potential utility of CVT-6883 is in several disease areas including asthma, chronic obstructive pulmonary disease, and pulmonary fibrosis [47, 48].
The discovery of three selective, high affinity A2B AdoR antagonists (10, 20, and 32) should aid in determining the pharmacological role of the A2B AdoR in various disease states in animal models and in clinical trials.
Abbreviations
- NECA:
-
5′-N-ethylcarboxamidoadenosine
- HEK:
-
human embryonic kidney cells
- cAMP:
-
cyclic adenosine monophosphate
- SAR:
-
structure-activity relationship
- dAUC:
-
dose-adjusted area under curve
- CHO:
-
Chinese hamster ovary
- BSMCs:
-
bronchial smooth muscle cells
- IL-6:
-
interleukin-6
- MCP-1:
-
monocyte chemotactic protein-1
- HLFs:
-
human lung fibroblasts
- OPN:
-
osteopontin
- MMPs:
-
matrix metalloproteases
References
Ryzhov S, Goldstein AE, Matafonov A, Zeng D, Biaggioni I, Feoktistov I (2004) Adenosine-activated mast cells induce IgE synthesis by B lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. J Immunol 172:7726–7733
Rorke S, Holgate ST (2002) Targeting adenosine receptors: novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Am J Respir Med 1:99–105
Akkari R, Burbiel JC, Hockemeyer J, Mueller CE (2006) Recent progress in the development of adenosine receptor ligands as antiinflammatory drugs. Curr Top Med Chem 6:1375–1399
Feokistov I, Polosa R, Hogate ST, Biaggioni I (1998) Adenosine A2B receptors: a novel therapeutic target in asthma? Trends Pharmacol Sci 19:148–153
Feoktistov I, Biaggioni I (1997) Adenosine A2B receptors. Pharmacol Rev 49:381–402
Fredholm BB, Ijzerman AP, Jacobson KA, Klotz K-N, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552
Holgate ST (2005) The identification of the adenosine A2B receptor as a novel therapeutic target in asthma. Br J Pharmacol 145:1009–1015
Zablocki J, Elzein E, Kalla R (2006) A2B adenosine receptor antagonists and their potential indications. Expert Opin Ther Patents 16:1347–1357
Cacciari B, Pastorin G, Bolcato C, Spalluto G, Bacilieri M, Moro S (2005) A2B adenosine receptor antagonists: recent developments. Mini Rev Med Chem 5:1053–1060
Baraldi PG, Aghazadeh M, Tabrizi A, Gessi S, Borea PA (2008) Adenosine receptor antagonists: translating medicinal chemistry and pharmacology into clinical utility. Chem Rev 108:238–263
Volpini R, Costanzi S, Vittori S, Cristalli G, Koltz K-N (2003) Medicinal chemistry and pharmacology of A2B adenosine receptors. Curr Top Med Chem 3:427–443
Jacobson KA, Ijzerman AP, Linden J (1999) 1,3-Dialkylxanthine derivatives having high potency as antagonists at human A2B adenosine receptors. Drug Dev Res 47:45–53
Suzuki F, Nonaka H, Ishii A (1992) 8-Polycycloalkyl-1,3-dipropylxanthines as potent and selective antagonists for A1-adenosine receptors. J Med Chem 35:924–930
Kim Y-C, Karton Y, Ji X-D, Melman N, Linden J, Jacobson KA (1999) Acyl-hydrazide derivatives of a xanthine carboxylic congener (XCC) as selective antagonists at human A2B adenosine receptors. Drug Dev Res 47:178–188
Daly JW, Padgett W, Shamim MT, Butts-Lamb P, Waters J (1985) 1,3-Dialkyl-8-(p-sulfophenyl)xanthines: potent water-soluble antagonists for A1- and A2-adenosine receptors. J Med Chem 28:487–492
Kim S-A, Marshall MA, Melman N, Kim HS, Muller CE, Linden J, Jacobson KA (2002) Structure-activity relationships at human and rat A2B adenosine receptors of xanthine derivatives substituted at the 1-, 3-, 7-, and 8-positions. J Med Chem 45:2131–2138
Hayallah AM, Sandoval-Ramirez J, Reith U, Schobert U, Preiss B, Schumacher B, Daly JW, Müller CE (2002) 1,8-Disubstituted xanthine derivatives: synthesis of potent A2B-selective adenosine receptor antagonists. J Med Chem 45:1500–1510
Zablocki J, Kalla R, Perry T, Palle V, Varkhedkar V, Xiao D, Piscopio A, Maa T, Gimbel A, Hao J, Chu N, Leung K, Zeng D (2005) The discovery of a selective, high affinity A2B adenosine receptor antagonist for the potential treatment of asthma. Bioorg Med Chem Lett 15:609–612
Kalla R, Perry T, Elzein E, Varkhedkar V, Li X, Ibrahim P, Palle V, Xiao D, Zablocki J (2004) A2B adenosine receptor antagonists, US Patent 6,825,349, 30 Nov 2004
Kalla R, Perry T, Elzein E, Varkhedkar V, Li X, Ibrahim P, Palle V, Xiao D, Zablocki J (2003) A2B adenosine receptor antagonists. WO Patent 2003/042214, 22 May 2003
Kalla R, Elzein E, Perry T, Li X, Palle V, Varkhedkar V, Gimbel A, Maa T, Zeng D, Zablocki J (2006) Novel 1,3-disubstituted 8-(1-benzyl-1H-pyrazol-4-yl) xanthines: high affinity and selective A2B adenosine receptor antagonists. J Med Chem 49:3682–3692
Elzein E, Kalla RV, Li X, Perry T, Gimbel A, Zeng D, Lustig D, Leung K, Zablocki J (2008) Discovery of a novel A2B adenosine receptor antagonist as a clinical candidate for chronic inflammatory airway diseases. J Med Chem 51:2267–2278
Kalla R, Elzein E, Perry T, Li X, Gimbel A, Yang M, Zeng D, Zablocki J (2008) Selective, high affinity A2B adenosine receptor antagonists: N-1 monosubstituted 8-(pyrazol-4-yl)xanthines. Bioorg Med Chem Lett 18:1397–1401
Elzein E, Kalla R, Li X, Perry T, Parkhill E, Palle V, Varkhedkar V, Gimbel A, Zeng D, Lustig D, Leung K, Zablocki J (2006) Novel 1,3-dipropyl-8-(1-heteroarylmethyl-1H-pyrazol-4-yl)-xanthine derivatives as high affinity and selective A2B adenosine receptor antagonists. Bioorg Med Chem Lett 16:302–306
Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D (2004) A2B adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol 30:118–125
Baraldi PG, Borea PA (2003) 8-Heteroaryl xanthine adenosine A2B receptor antagonists. WO Patent 2003/063800, 7 Aug 2003
Baraldi PG, Tabrizi MA, Preti D, Bovero A, Romagnoli R, Fruttarolo F, Zaid NA, Moorman AR, Varani K, Gessi S, Merighi S, Borea PA (2004) Design, synthesis, and biological evaluation of new 8-heterocyclic xanthine derivatives as highly potent and selective human A2B adenosine receptor antagonists. J Med Chem 47:1434–1447
Baraldi PG, Tabrizi MA, Preti D, Bovero A, Fruttarolo F, Romagnoli R, Moorman AR, Gessi S, Merighi S, Varani K, Borea PA (2004) [3H]-MRE 2029-F20, a selective antagonist radioligand for the human A2B adenosine receptors. Bioorg Med Chem Lett 14:3607–3610
Wang G, Rieger JM, Thompson RD (2006) Pyridyl substituted xanthines. WO Patent 2006/091896, 31 Aug 2006
Grahner B, Winiwarter S, Lanzner W, Müller CE (1994) Synthesis and structure-activity relationships of deazaxanthines: analogs of potent A1- and A2-adenosine receptor antagonists. J Med Chem 37:1526–1534
Esteve C, Nueda A, Diaz JL, Beleta J, Cardenas A, Lozoya E, Cadavid MI, Loza MI, Ryder H, Vidal B (2006) New pyrrolopyrimid-6-yl benzenesulfonamides: potent A2B adenosine receptor antagonists. Bioorg Med Chem Lett 16:3642–3645
Stefanchi A, Brea JM, Cadavid MI, Centeno NB, Esteve C, Loza MI, Martinez A, Nieto R, Raviña E, Sanz F, Segarra V, Sotelo E, Vidal B, Carotti A (2008) 1-, 3- and 8-substituted-9-deazaxanthines as potent and selective antagonists at the human A2B adenosine receptor. Bioorg Med Chem 16:2852–2869
Kalla R, Elzein E, Marquart T, Perry T, Li X, Zablocki J (2005) A2B adenosine receptor antagonists. WO Patent 2005/042534, 12 May 2005
Vidal B, Eastwood PR, Rodriguez JG (2005) Condensed pyridine derivatives useful as A2B adenosine receptor antagonists. WO Patent 2005/100353, 27 Oct 2005
Vidal B, Trias CE (2005) Pyrimidin-2-amine derivatives and their use as A2B adenosine receptor antagonists. WO Patent 2005/040155, 6 May 2005
Vidal B, Nueda A, Esteve C, Domenech T, Benito S, Reinoso RF, Pont M, Calbet M, López R, Cadavid MI, Loza MI, Córdenas A, Godessart N, Beleta J, Warrellow G, Ryder H (2007) Discovery and characterization of 4′-2(2-furyl)-N-pyridin-3-yl-4,5′-bipyrimidin-2′-amine (LAS38096), a potent, selective, and efficacious A2B adenosine receptor antagonist. J Med Chem 50:2732–2736
Müller CE, Stein B (1996) Adenosine receptor antagonists: structure and potential therapeutic applications. Curr Pharm Des 2:501–530
Thomson RD, Secunda S, Daly JW, Olsson RA (1991) N 6, 9-Disubstituted adenines: potent, selective antagonists at the A1 adenosine receptor. J Med Chem 34:2877–2882
Camaioni E, Costanzi S, Vittori S, Volpini R, Klotz K-N, Cristalli G (1998) New substituted 9-alkylpurines as adenosine receptor ligands. Bioorg Med Chem 6:523–533
Harada H, Asano O, Hoshino Y, Yoshikawa S, Matsukura M, Kabasawa Y, Niijima J, Kotake Y, Watanabe N, Kawata T, Inoue T, Horizoe T, Yasuda N, Minami H, Nagata K, Murakami M, Nagaoka J, Kobayashi S, Tanaka I, Abe S (2001) 2-Alkynyl-8-aryl-9-methyladenines as novel adenosine receptor antagonists: their synthesis and structure-activity relationships toward hepatic glucose production induced via agonism of the A2B receptor. J Med Chem 44:170–179
Harada H, Asano O, Kawata T, Inoue T, Horizoe T, Yasuda N, Nagata K, Murakami M, Nagaoka J, Kobayashi S, Tanaka I, Abe S (2001) 2-Alkynyl-8-aryladenines possessing an amide moiety: their synthesis and structure-activity relationships of effects on hepatic glucose production induced via agonism of the A2B adenosine receptor. Bioorg Med Chem 9:2709–2726
Stewart M, Steinig AG, Ma C, Song J-P, McKibben B, Castelhano AL, MacLennan SJ (2004) [3H]OSIP339391, a selective, novel, and high affinity antagonist radioligand for adenosine A2B receptors. Biochem Pharmacol 68:305–312
Feoktistov I, Ryzhov S, Zhong H, Goldstein AE, Matafonov A, Zeng D, Biaggioni I (2004) Hypoxia modulates adenosine receptors in human endothelial and smooth muscle cells toward an A2B angiogenic phenotype. Hypertension 44:649–654
Zhong H, Belardinelli L, Maa T, Zeng D (2005) Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol 32:2–8
Gessi S, Varani K, Merighi S, Cattabriga E, Pancaldi C, Szabadkai Y, Rizzuto R, Klotz K-N, Leung E, Mac Lennan S, Baraldi PG, Borea PA (2005) Expression, pharmacological profile, and functional coupling of A2B receptors in a recombinant system and in peripheral blood cells using a novel selective antagonist radioligand, [3H]MRE 2029-F20. Mol Pharmacol 67:2137–2147
CV Therapeutics press release, www.cvt.com
Sun C-X, Zhong H, Mohsenin A, Morschi E, Chunn JL, Molina JG, Belardinell L, Zeng D, Blackburn M (2006) Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest 116:2173–2182
Mustafa SJ, Nadeem A, Fan M, Zhong H, Belardinelli L, Zeng D (2007) Effect of a specific and selective A2B adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J Pharmacol Exp Ther 320:1246–1251
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kalla, R.V., Zablocki, J. Progress in the discovery of selective, high affinity A2B adenosine receptor antagonists as clinical candidates. Purinergic Signalling 5, 21–29 (2009). https://doi.org/10.1007/s11302-008-9119-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-008-9119-x