
Abstract
Allosteric modulators for adenosine receptors may have potential therapeutic advantage over orthosteric ligands. Allosteric enhancers at the adenosine A1 receptor have been linked to antiarrhythmic and antilipolytic activity. They may also have therapeutic potential as analgesics and neuroprotective agents. A3 allosteric enhancers are postulated to be useful against ischemic conditions or as antitumor agents. In this review, we address recent developments regarding the medicinal chemistry of such compounds. Most efforts have been and are directed toward adenosine A1 and A3 receptors, whereas limited or no information is available for A2A and A2B receptors. We also discuss some findings, mostly receptor mutation studies, regarding localization of the allosteric binding sites on the receptors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The superfamily of human G protein-coupled receptors (GPCRs) consists of three major classes: A, B, and C. Class A, also termed rhodopsin-like after the prototypic visual pigment, constitutes the largest group of GPCRs. Adenosine receptors belong to this class. Four subtypes of adenosine receptors have been cloned and pharmacologically characterized: A1, A2A, A2B, and A3. Among the human adenosine receptors, the most similar are A1 and A3 (49% sequence similarity) and A2A and A2B (59% similarity). Activation of A1 and A3 receptors induces the inhibition of the enzyme adenylate cyclase mediated by an inhibitory G protein (Gi). Activation of A2A and A2B receptors leads to a Gs-mediated stimulation of this enzyme [1].
Many representatives of class A receptors, among them adenosine receptors, have been shown to be allosterically regulated. Allosteric refers to binding sites that are different from the orthosteric primary ligand-binding site to which ligands such as endogenous adenosine and synthetic derivatives thereof bind. The binding of modulators is supposed to result in conformational changes of the receptor that affect binding of the orthosteric ligand with consequences for receptor activity (see Fig. 1).

Binding of an allosteric ligand (1) modulates the binding of hormone or neurotransmitter (2)
Strictly speaking, the allosteric modulator in this scenario has no activity by itself and needs the orthosteric ligand (often the endogenous hormone or neurotransmitter) in order to become activated. The nature of allosteric modulation is therefore very much dependent on this orthosteric ligand. As a consequence, the overall pharmacology invoked by the allosteric modulator resembles physiology more closely than is possible with most of today’s drugs, i.e., synthetic orthosteric ligands (see Fig. 2 for the effect of an allosteric enhancer).

Allosteric modulators may either increase or decrease the effect of an agonist or an antagonist. An allosteric enhancer can be a more physiologic alternative to synthetic agonists (b, c). An allosteric enhancer can only act in the presence of an (endogenous) agonist. It mimics the duration and intensity of action of a hormone or neurotransmitter (a) much better than a synthetic agonist does (c). This combined action of two compounds (i.e., agonist and enhancer) might also induce selectivity of drug action
The concept of allosteric modulation of established drug targets such as GPCRs has fueled an intensive quest for new classes of lead compounds different from classic agonists and antagonists. This has been the subject of several recent reviews [2–5]. As adenosine receptors were among the first GPCRs found to be allosterically regulated, these receptors have often been reviewed also [6–8]. The aim of this review is to summarize the latest developments in this fascinating field for all four subtypes of adenosine receptors, with a twofold emphasis. First, chemical classes of allosteric ligands, their receptor preference, and structure-activity relationships (SAR) are discussed. Second, attention is given to the putative binding site of the modulators.
A1 adenosine receptors
Baraldi et al. recently published an extensive review of allosteric modulators of A1 receptors [8]. Here, we briefly provide an overview of historical as well as new allosteric modulators for A1 adenosine receptors. Bruns and coworkers introduced the first allosteric modulators of the A1 receptor in 1990 [9, 10]. Three compounds, (2-amino-4,5,6,7-tetrahydro-benzo[b]thiophen-3-yl)-(2-chloro-phenyl)-methanone (PD 71,605); 2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]methanone (PD 81,723); and (2-amino-6-benzyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridin-3-yl)(4-chloro-phenyl)methanone (PD 117,975) were studied in detail (see Fig. 3). These chemical structures were part of the Parke-Davis compound library at the time and were originally used as intermediates in the synthesis of benzodiazepine-like compounds [11].

2-Amino-3-substituted thiophene derivatives, allosteric modulators for A1 adenosine receptors
Their allosteric-enhancing nature was identified by accident while screening the library in a radioligand-binding assay at the rat A1 adenosine receptor. The three compounds increased rather than decreased (the original aim of the screening campaign) the binding of the agonist radioligand [3H]N6-cyclohexyladenosine to A1 receptors in rat-brain membranes. The enhancers appeared to slow the dissociation of the agonist [3H]N6-cyclohexyladenosine from the A1 receptor, indicating an allosteric mechanism of action. These compounds were also competitive antagonists at the same receptor. Therefore, the concentration range within which these compounds enhance the effects of agonists is limited. Among the compounds tested by Bruns et al., PD 81,723 stood out as one of the more potent compounds with the best ratio of enhancing to antagonistic action. PD 81,723 has been thoroughly and independently studied over the years, and it became the reference allosteric modulator of A1 receptors. It has been extensively modified structurally [9, 10].
Van der Klein and colleagues synthesized a new series of 2-amino-3-benzoylthiophenes with PD 81,723 as a template. Among these compounds, 1–3 (Fig. 3) proved to be more potent enhancers than PD 81,723, with comparable antagonistic activity. The biological assays were carried out on membranes of rat-brain cortex, and the enhancing activity was measured at 10 μM of test compound. For compound 1, enhancement was 122%; for compound 2, 123%; and for compound 3, 151% compared with that of PD 81,723 (100%).
It was concluded that in this class of 2-amino-3-benzoylthiophene derivatives, lipophilic benzoyl (3-CF3, 3,4-Cl, or 4-t-butyl) substitution and thiophene 4-alkyl substitution favored allosteric enhancing activity. Thiophene 5-bulky substitution favored antagonistic properties, which rendered the separation of these two activities in these structures possible [12, 13].
Baraldi et al. also developed a series of PD 81,723 analogues. For their pharmacological evaluation, cyclic adenosine monophosphate (cAMP) determinations were carried out in Chinese hamster ovary (CHO) cells expressing human A1 receptors (CHO:hA1) vs. radioligand dissociation experiments on rat-brain membranes. In their studies, compounds 2, 4, and 5 (Fig. 3) proved to be nearly as efficacious (effect on Emax) as and more potent (effect on EC50) than PD 81,723 and caused a significant reduction of the cAMP content of CHO:hA1 cells at a concentration of 0.1 μM [14]. In a later and more extensive study, the same authors investigated a large number of 2-amino-3-naphthoylthiophenes [15]. Among these compounds, 6 was the most active, increasing agonist binding to CHO:hA1, human-brain, and rat-cortex membranes by 149%, 43%, and 27% (51%, 15%, and 22% for PD 81,723). In the series of 4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivatives, the only compound that showed significant allosteric enhancer activity at any concentration was compound 7. A series of 2-amino-3-heteroarylthiophenes have also been synthesized. The same functional cAMP assay also revealed that compounds 8–11 are allosteric enhancers at the human A1 receptor. Replacement of the phenyl ring at the 3 position of the thiophene ring with a 2-or 3-thienyl moiety maintained the enhancing activity of PD 81,723 [16]. Thieno[2,3-c]pyridine derivatives were accessed by the Baraldi group by means of microwave-assisted synthesis. Among the prepared compounds, 12 showed activity comparable with that of PD 81,723 (Fig. 3) [17].
Tranberg et al. also performed a systematic survey of the PD 81,723 lead structure. The 3-OMe derivative 13 had high enhancing activity and relatively low antagonistic potency: 99% and 13%, respectively, at 100 μM [18]. The same research group showed that chemical structures more different from the prototypic PD 81,723 were allosteric enhancers also [19]. The allosteric enhancing activity score of PD 81,723 was 19% and served as a standard for comparison. For compound 14, it was 48%; and for the 3-(1-naphthoyl substituted compound 15, it was 52%. Compounds 16 and 17 seemed to be more potent, with scores of 91% and 80%, respectively (Fig. 3). In this study assessment of allosteric enhancing activity was done in a somewhat different way and consisted of three phases: (1) binding to equilibrium of the agonist N6–4-amino-3-iodo-benzyladenosine (125I-ABA) to the A1 adenosine receptor–G protein ternary complex, (2) stabilization of that complex by the allosteric enhancer, and (3) dissociation of the complex by adding a combination of an A1 receptor antagonist and guanosine 5’-[γ-thio]triphosphate (GTPγS), to accelerate agonist radioligand dissociation. A score of 100% means no dissociation and a score of zero complete dissociation. Similarly, Nikolakopoulos et al. studied 2-aminothiophene-3-carboxylates and carboxamides as adenosine A1 receptor allosteric enhancers. Compound 18 was notably more potent and efficacious than PD 81,723 (Fig. 3) [20]. Interestingly, 2-aminoselenophene-3-carboxylates also proved to be allosteric enhancers of A1 receptors [21].
In 2006, the first radiolabeled adenosine A1 allo-steric enhancer, the tritiated form of compound 2, was prepared (Fig. 3). The biological investigation of this compound, 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl)-(4-chlorophenyl)-methanone ([3H]T-62), suggested the presence of an allosteric binding site on the adenosine A1 receptor distinct from the orthosteric binding site for agonists and antagonists [22].
Another chemical class of allosteric enhancers of agonist binding at cloned hA1 adenosine receptors are the 2-aminothiazoles. The most potent and efficacious enhancer of the series reported is compound 19 (Fig. 4). The potency of this 2-aminothiazole derivative was reported to be 127 times that of PD 81,723 (EC50 values 0.3 μM and 38 μM, respectively). The intrinsic activity of compound 19 was about twice that of PD 81,723. The enhancing effect at A2A and A3 receptors by this compound was found to be weak [23].

2-Aminothiazoles and [1,2,4]thiadiazole analogues, questionable allosteric modulators for A1 adenosine receptors
We also prepared a number of 2-aminothiazoles and their amide derivatives. The 2-aminothiazoles (the free bases) were tested as allosteric enhancers of agonist binding to human A1 receptors. In a variety of experimental setups, the compounds showed no such effect, in contrast to the above findings of Chordia et al. [23]. Some of the amides emerged as moderately active antagonists on human A1 and/or A2A receptors, with lower or negligible potency at A3 receptors [24].
Chordia et al. next investigated these contrasting findings in more detail and concluded that the free bases of this type of compound in fresh solutions are only weakly active as allosteric enhancers. However, the allosteric enhancing activity in solutions of these compounds in dimethylformamide (DMF), dimethylsulfoxide (DMSO), or acetonitrile increased after 4 h and finally led to a constant activity equivalent to that of the hydriodide salts after 12 h [25].
Fawzi et al. reported SCH-202676 (N-(2,3-diphenyl-[1,2,4]thiadiazole-5(2H)-ylidene)methanamine) to act as a more promiscuous agent (Fig. 4). It appeared to modulate the activity of many GPCRs, including opioid, muscarinic, adrenergic, and dopaminergic receptors [26]. We studied its activity on adenosine receptors. We also synthesized a number of 2,3,5-substituted [1,2,4]thiadiazole analogues of SCH-202676 and tested these as potential allosteric modulators of adenosine receptors. SCH-202676 displayed a mixed behavior on adenosine receptors. It displaced agonist binding to A1 receptors in a reversible manner and both agonist and antagonist binding to A2A receptors. The displacement curves were very steep, dissimilar to normal competitive displacement curves. On A1 receptors, compound 20 was shown to be an allosteric enhancer of antagonist 8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX) binding (Fig. 4) [27].
In our next work, we investigated a new series of 2,3,5-substituted [1,2,4]thiadiazoles. Results of receptor-ligand-binding experiments and stability studies by high-performance liquid chromatography (HPLC) and HPLC mass spectrometry (MS) showed that the compounds are highly reactive sulfhydryl-modifying agents rather than allosteric inhibitors. They appear to be reduced into their thiourea precursors by thiol groups of, e.g., cysteine residues in proteins. This general feature most probably explains their overall lack of selectivity [28].
Amiloride (a diuretic) also acts as a promiscuous agent, which had been reported before to interact with many GPCRs such as α2-adrenergic receptors [29]. Results by Garritsen et al. suggest that amiloride interacts with the A1 receptor at a site distinct from the ligand-binding site. Furthermore, amiloride appeared to be an A1 receptor antagonist with binding characteristics that differ from the classic A1 receptor antagonists such as DPCPX [30–31].
Allosteric modulation of A1 receptors has been demonstrated not only in vitro but also in vivo in various species, including humans. Local administration of PD 81,723 in rats caused a concentration-dependent increase of extracellular acetylcholine levels of approximately 40%. However, the antagonistic actions of the allosteric enhancer PD 81,723 on the adenosine A1 receptor canceled the allosteric actions [32]. Treating dogs with PD 81,723 reduced the time required for ischemia to produce preconditioning [33]. Compound 2 (T-62) has been in preclinical development for the treatment of chronic pain [34, 35] and is now in phase II clinical trials (King Pharmaceuticals).
A2A adenosine receptors
Although an allosteric enhancer for the A2A receptors has not yet been developed, amiloride and analogues were demonstrated to be allosteric inhibitors also for the A2A receptor. Among the derivatives tested, 5-(N,N-hexamethylene)amiloride (HMA) proved to be the most potent allosteric inhibitor (Fig. 5). Amiloride analogues increased the dissociation rate of the antagonist 4-{2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a]1,3,5]triazin-5-yl-amino]ethyl}phenol ([3H]ZM241385) from the A2A receptor. However they did not show any effect on the dissociation rate of the agonist 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5’-N-ethylcarboxamidoadenosine ([3H]CGS21680) [36]. In contrast to the effects of amiloride analogues on the A2A receptor, sodium ions decreased the dissociation rate of the antagonist [3H]ZM241385 from the A2A receptor in a concentration-dependent manner.

Amiloride derivatives, allosteric modulators of adenosine receptors
Gao and collegues later showed that amiloride analogues are also allosteric inhibitors of antagonist binding at A1 and A3 receptor subtypes [37]. Amiloride and 5-(N,N-dimethyl)amiloride (DMA) were more potent at A1 receptors than at A3 receptors, whereas HMA was more potent at A3 receptors (Fig. 5).
A2B adenosine receptors
Allosteric modulators for A2B receptors have not yet been described. Research on the A2B receptor subtype has been hampered by the lack of suitable, commercially available radioligands. However, with the advent of the radiolabeled antagonist 8-(4-[{(4-cyanophenyl)carbamoylmethyl}oxy]phenyl)-1,3-di(n-propyl)xanthine ([3H]MRS1754) and other radioligands, such studies are now feasible. Still, a radiolabeled agonist is not yet available, which is hindering the identification of allosteric enhancers of agonist binding.
A3 adenosine receptors
Gao et al. reported the first selective allosteric enhancers of agonist binding at human A3 receptors [38]. The effects of the reference A3 receptor agonist 2-chloro-N6-(3-iodobenzyl)-5’-N-methylcarboxamidoadenosine (Cl-IB-MECA) on forskolin-induced cAMP formation were significantly enhanced by several 3-(2-pyridinyl)isoquinoline derivatives. Most compounds, however, also displayed orthosteric binding affinity to the human A3 receptor. Their results indicated that 4-methoxy-N-[7-methyl-3-(2-pyridinyl)-1-isoquinolinyl]benzamide (VUF5455) (see Fig. 6) is selective for the agonistic state of the A3 receptor. In competitive binding studies on cloned human A3 receptors VUF5455 is rather weak as an orthosteric ligand (Ki = 1680 nM). Replacement of the 7-methyl group of VUF5455 by H [4-methoxy-N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide (VUF8504)] had no influence on the allosteric activity but increased the A3 receptor affinity nearly 100-fold (Ki = 17.3 nM). Exchanging the 4’-methoxy group of VUF8504 by methyl [4-methyl-N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide (VUF8502)] or H [N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide (VUF8507)] lowered the A3 receptor affinity (Ki values of 96 nM and 204 nM, respectively) without affecting the allosteric activity. The corresponding imino instead of carboxamido analogues displayed moderate A3 affinity (Ki values 300–700 nM) but were devoid of allosteric properties. Thus, although VUF5455 is not devoid of A3 receptor antagonistic activity, the compound might be used as a lead for the design of pure allosteric enhancers of A3 receptors.

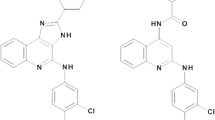
Structures of allosteric modulators of A3 receptors
Van Galen et al. identified 1H-imidazo-[4,5-c]quinolines as nonxanthine adenosine receptor antagonists [39]. Later, these types of compounds were shown to represent a second series of allosteric modulators for the A3 receptor. Among these compounds, 2-cyclopentyl-4-phenylamino-1H-imidazo[4,5-c]quinoline (DU124183) (see Fig. 6) selectively enhanced agonist binding and function at A3 receptors, similar to VUF5455. However, in contrast to VUF5455, DU124183 also increased the maximum efficacy of A3 receptor agonists in the cAMP assay with cloned human A3 receptors. For DU124183, there was a marked leftward shift of the concentration-response curve of the A3 receptor agonists in the presence of an orthosteric antagonist and a potentiation of the maximum agonist efficacy by approximately 30%. In competitive binding studies, DU124183 also displayed some orthosteric activity, as it bound with moderate affinity (Ki = 820 nM) to human A3 receptors. Some members of this series also had a competitive interaction with the A1 adenosine receptor [39, 40].
The aim of a subsequent study was the optimization of DU124183. Structural modifications were made at the 4-amino and 2 positions of the molecule, partly using the Topliss scheme [41]. The compounds were tested in binding and functional assays (Table 1). SARs for the allosteric enhancing effects showed that at the 4-amino position, a phenyl ring or substituted monocyclic phenyl ring (compounds DU124183, 21) was more enhancing than bicyclic aryl rings (compounds 22, 25). A phenyl group (DU124183) was also better in enhancing than the corresponding benzyl and phenylethyl groups (24, 25). As noted previously, substitution at the 2-position was also necessary for allosteric enhancement (compound 29). At this position, medium-sized cycloalkyl substituents (cyclopentyl and cyclohexyl) were most favorable for enhancement. Analogues bearing smaller or larger rings (compounds 28, 26) or an acyclic alkyl group (compound 30) were considerably less enhancing, and similarly an aromatic five-membered ring (compound 27) was not conductive to enhancement. Two compounds, 2-cyclopentyl-4-benzylamino (24) and 2-cyclohexyl-4-(3,4-dichlorophenyl)amino (LUF6000, see Fig. 6) analogues potentiated the maximum efficacy of the agonist Cl-IB-MECA by 45–50% (Table 1). Moreover, LUF6000 enhanced agonist efficacy in a functional assay and decreased the agonist dissociation rate without influencing agonist potency. Most allosteric enhancers in this study only modestly inhibited orthosteric ligand binding, if at all. Apparently, structural requirements for allosteric enhancement at the A3 receptor are distinct from those for the inhibition of equilibrium radioligand binding. This suggests even greater selectivity is feasible in a future lead optimization program. Allosteric enhancers of A3 receptor activation are predicted to be useful against a number of disorders, including ischemic conditions and cancer. LUF6000 showed no effect on the basal receptor activity yet showed a significant enhancement of agonist action. This result suggests that such enhancers could be safer drugs and would be particularly effective to enhance agonist efficacy under pathophysiological conditions such as brain and heart ischemia, where adenosine and inosine concentrations are greatly increased [42; Gao et al., manuscript submitted 2008].
Mutagenesis studies, binding sites for allosteric modulators
The aim of several publications was to determine where the binding sites are for allosteric modulators on adenosine receptors. Theoretically, it might be possible that PD 81,723 binds to another protein, such as a G-protein subunit, and that this complex allosterically regulates the A1 receptor. However, Bhattacharya et al. suggested that PD 81,723 binds to the A1 receptor itself and that it stabilizes the high-affinity conformational state of the A1 receptor–G-protein complex [43].
Site-directed mutagenesis is a useful technique for elucidating ligand–receptor interactions by testing the role of several amino acids within the receptor. Mutant A1 receptors have been studied, where glycine at position 14 had been changed into threonine (G14T). The G14T mutant renders the receptor constitutively active with (very) high agonist affinity, upon which PD 81,723 loses its enhancing activity. It appears that the G14T receptor is “locked” in an active state and as such does not reveal the site of PD 81,723 binding [44]. Studies on wild-type and mutant (T277A) A1 receptors gave analogous results. Mutation of a threonine residue to alanine at position 277 in the human A1 receptor decreased agonist binding to the receptor but also abolished the effects of PD 81,723 [45]. Again, it would be premature to conclude that T277 is part of the PD 81,723 binding site.
Barbhaiya et al. mutated a largely conserved aspartic acid residue in TM2 of the A1 receptor to alanine (D55A). This residue was found to be responsible for allosteric regulation of ligand-A1 receptor binding by sodium ions [46]. Gao and colleagues performed mutagenesis experiments on the A2A receptor, where a glutamic acid residue in TM1 and a histidine residue in TM7 were mutated to glutamine (E13Q) and tyrosine (H278Y), respectively. Results suggest that the two closely linked residues Glu13 and His278 in the A2A receptor are the most important for agonist recognition and partly responsible for the allosteric regulation by sodium ions [36, 47]. Furthermore Gao et al. identified residues involved in the allosteric modulation of the human A3 receptor. A number of residues in the transmembrane helical domains were shown to be critically involved in the recognition of the allosteric modulators. The F182A (TM5) and N274A (TM7) mutations abolished the allosteric effects of the two modulators DU124183 and VUF5455 but had hardly any effect on agonist binding, as was the case for N30A (TM1) and D58N (TM2). The D107N (TM3) mutation eliminated the effects of DU124183 but not of VUF5455 [48].
To summarize, site-directed mutagenesis studies may shed light on amino acids involved in allosteric ligand-receptor interactions. Experiments to that end have been carried out on A1, A2A, and A3 receptors, whereas A2B receptors have not yet been studied. For A1 receptors, Asp55 in TM2 is probably responsible for allosteric regulation of binding by sodium ions and amilorides, whereas upon G14T (TM1) and T277A (TM7) mutagenesis, PD 81,723 loses its enhancing activity with respect to carboxypeptidase A (CPA). For A2A receptors, Glu13 in TM1 and His278 in TM7 have been shown to be linked to allosteric regulation by sodium ions. For A3 receptors, F182A (TM5) and N274A (TM7) mutations abolished the allosteric effects of the two modulators DU124183 and VUF5455. The D107N (TM3) mutation eliminated the effects of DU124183 but not of VUF5455. It is fair to assume that the residues studied so far only yield a fragmented view of the amino acids involved in the allosteric regulation of adenosine receptors. The location of the residues discussed here is shown in a receptor homology model of the adenosine receptor based on the recently reported crystal structure of the β2-adrenergic receptor [49] (Fig. 7).

The location of amino acid residues involved in allosteric modulation of A1, A2A, and A3 receptors as displayed in a receptor homology model of the adenosine receptor (side and top view) based on the structure of the β2-adrenergic receptor cocrystallized with its ligand carazolol (PDB coordinates: 2rh1). C-α atoms at these locations are shown as spheres. The position of D55 in the A1 receptor is equivalent to D58 in the A3 receptor
Conclusions
Allosteric modulation is a new way of intervening with receptor function. Because of the ability of allosteric modulators to modulate receptor conformations in the presence of orthosteric ligand, allosteric modulators can “fine tune” classical pharmacological responses. Although an allosteric modulator may not possess efficacy by itself, it can provide a powerful therapeutic advantage over orthosteric ligands, namely, the ability to selectively influence tissue responses only when the endogenous agonist is present.
Abbreviations
- AR:
-
adenosine receptor
- cAMP:
-
cyclic adenosine monophosphate
- cDNA:
-
complementary deoxyribonucleic acid
- CGS21680:
-
2-[p-(2-carboxyethyl)phenyl-ethylamino]-5’-N-ethylcarboxamidoadenosine
- CGS15943:
-
5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline
- CHO:
-
Chinese hamster ovary
- Cl-IB-MECA:
-
2-chloro-N6-(3-iodobenzyl)-5’-N-methylcarboxamidoadenosine
- CP:
-
cyclopentyl
- DMA:
-
5-(N,N-dimethyl)amiloride
- DMF:
-
dimethylformamide
- DMSO:
-
dimethylsulfoxide
- DPCPX:
-
8-cyclopentyl-1,3-dipropylxanthine
- DU124183:
-
2-cyclopentyl-4-phenylamino-1H-imidazo[4,5-c]quinoline
- GPCR:
-
G protein-coupled receptor
- GTPγS:
-
guanosine 5’-[γ-thio]triphosphate
- h:
-
human
- HMA:
-
5-(N,N-hexamethylene)amiloride
- HPLC-MS:
-
high-performance liquid chromatography mass spectrometry
- I-ABA:
-
N6–4-amino-3-iodo-benzyladenosine
- I-AB-MECA:
-
N6-(4-amino-3-iodobenzyl)-5’-N-methylcarbamoyladenosine
- LUF6000:
-
N-(3,4-dichlorophenyl)-2-cyclohexyl-1H-imidazo[4,5-c]quinolin-4-amine
- MRS1754:
-
8-(4-[{(4-cyanophenyl)carbamoylmethyl}oxy]phenyl)-1,3-di(n-propyl)xanthine
- NECA:
-
5’-N-ethylcarboxamidoadenosine
- PD 71,605:
-
(2-amino-4,5,6,7-tetrahydro-benzo[b]thiophen-3-yl)-(2-chloro-phenyl)-methanone
- PD 117,975:
-
(2-amino-6-benzyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridin-3-yl)(4-chloro-phenyl)methanone
- PD 81,723:
-
2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]methanone
- R-PIA:
-
N6-[(R)-phenylisopropyl]adenosine
- SAR:
-
structure-activity relationships
- SCH-202676:
-
(N-(2,3-diphenyl-[1,2,4]thiadiazole-5(2H)-ylidene)methanamine)
- T-62:
-
(2-amino-4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl)-(4-chlorophenyl)-methanone
- TM:
-
transmembrane domain
- VUF5455:
-
4-methoxy-N-[7-methyl-3-(2-pyridinyl)-1-isoquinolinyl]benzamide
- VUF8502:
-
4-methyl-N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide
- VUF8504:
-
4-methoxy-N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide
- VUF8507:
-
N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide
- ZM241385:
-
4-{2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a]1,3,5]triazin-5-yl-amino]ethyl}phenol
References
Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J (2001) Pharmacol Rev 53:527–552
Soudijn W, van Wijngaarden I, IJzerman AP (2004) Allosteric modulation of G protein-coupled receptors: perspectives and recent developments. Drug Discov Today 9:752–758
Soudijn W, van Wijngaarden I, IJzerman AP (2002) Allosteric modulation of G protein-coupled receptors. Curr Opin Drug Discov Devel 5:749–755
May LT, Leach K, Sexton PM, Christopoulos A (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51
Gao Z-G, Jacobson KA (2006) Allosterism in membrane receptors. Drug Discov Today 11:191–202
Jacobson KA, Gao Z-G (2006) Adenosine receptors as therapeutic targets. Nature Rev Drug Discov 5:247–264
Gao Z-G, Kim S-K, IJzerman AP, Jacobson KA (2005) Allosteric modulation of the adenosine family of receptors. Mini Rev Med Chem 5:545–553
Baraldi PG, Iaconinoto MA, Moorman AR, Carrion MD, Cara CL, Preti D, Lopez OC, Fruttarolo F, Tabrizi MA, Romagnoli R (2007) Allosteric enhancers for A1 adenosine receptor. Mini Rev Med Chem 7:559–569
Bruns RF, Fergus JH, Coughenour LL, Courtland GG, Pugsley TA, Dodd JH, Tinney FJ (1990) Structure-activity relationships for enhancement of adenosine A1 receptor binding by 2-amino-3-benzoylthiophenes. Mol Pharmacol 38:950–958
Bruns RF, Fergus JH (1990) Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol 38:939–949
Tinney FJ, Sanchez JP, Nogas JA (1974) Synthesis and pharmacological evaluation of 2,3-dihydro-1H-thieno[2,3-e][1,4]diazepines. J Med Chem 17:624–630
van der Klein PAM, Kourounakis AP, IJzerman AP (1999) Allosteric modulation of the adenosine A1 receptor: Synthesis and biological evaluation of novel 2-amino-3-benzoylthiophenes as allosteric enhancers of agonist binding. J Med Chem 42:3629–3635
Kourounakis AP, van de Klein PAM, IJzerman AP (2000) Elucidation of stucture-activity relationships of 2-amino-3-benzoylthiophenes: study of their allosteric enhanceing vs. antagonistic activity on adenosine A1 receptors. Drug Dev Res 49:227–237
Baraldi PG, Zaid AN, Lampronti I, Fruttarolo F, Pavani MG, Tabrizi MA, Shryock JC, Leung E, Romagnoli R (2000) Synthesis and biological effects of a new series of 2-amino-3benzoylthiophenes as allosteric enhancers of A1 adenosine receptor. Bioorg Med Chem Lett 10:1953–1957
Baraldi PG, Romagnoli R, Pavani MG, Nunez MC, Tabrizi MA, Shryock JC, Leung E, Moorman AR, Uluoglu C, Iannotta V, Merighi S, Borea PA (2003) Synthesis and biological effects of novel 2-amino-3-naphthoylthiophenes as allosteric enhancers of the A1 adenosine receptor. J Med Chem 46:794–809
Baraldi PG, Pavani MG, Shryock JC, Moorman AR, Iannatta V, Borea PA, Romagnoli R (2004) Synthesis of 2-amino-3-heteroaroylthiophenes and evaluation of their activity as potential allosteric enhancers at the human A1 receptor. Eur J Med Chem 39:855–865
Romagnoli R, Baraldi PG, Moorman AR, Iaconinoto MA, Carrion MD, Cara CL, Tabrizi MA, Preti D, Fruttarolo F, Baker SP, Varani K, Borea PA (2006) Microwave-assisted synthesis of thieno[2,3-c]pyridine derivatives as a new series of allosteric enhancers at the adenosine A1 receptor. Bioorg Med Chem 16:5530–5533
Tranberg CE, Zickgraf A, Giunta BN, Lütjens H, Figler H, Merphree LJ, Falke R, Fleischer H, Linden J, Scammels PJ, Olsson RA (2002) 2-Amino-3-aroyl-4,5-alkylthiopenes: Agonist allosteric enhancers at human A1 adenosine receptors. J Med Chem 45:382–389
Lütjens H, Zickgraf A, Figler H, Linden J, Olsson RA, Scammels PJ (2003) 2-Amino-3-benzoylthiophene allosteric enhancers of A1 adenosine agonist binding: New 3-, 4-, and 5-modifications. J Med Chem 46:1870–1877
Nikolakopoulos G, Figler H, Linden J, Scammels PJ (2006) 2-Aminothiophene-3-carboxylates and carboxamides as adenosine A1 receptor allosteric enhancers. Bioorg Med Chem 14:2358–2365
Aumann KM, Scammels PJ, White JM, Schiesser CH (2007) On the stability of 2-aminoselenophene-3-carboxylates: potential dual-acting selenium-containing allosteric enhancers of A1 adenosine receptor binding. Org Biomol Chem 5:1276–1281
Romagnoli R (2006) Synthesis and biological characterization of [3H](2-amino-4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl)-(4-chlorophenyl)-methanone, the first radiolabelled adenosine A1 allosteric enhancer. Bioorg Med Chem 16:1402–1404
Chordia MD, Murphree LJ, Macdonald TL, Linden J, Olsson RA (2002) 2-Aminothiazoles: A new class of agonist allosteric enhancers of A1 adenosine receptors. Bioorg Med Chem Lett 12:1563–1566
Göblyös A, Santiago SN, Pietra D, Mulder-Krieger T, von Frijtag Drabbe Künzel J, Brussee J, IJzerman AP (2005) Synthesis and biological evaluation of 2-aminothiazoles and their amide derivatives on human adenosine receptors. Lack of effect of 2-aminothiazoles as allosteric enhancers. Bioorg Med Chem 13:2079–2087
Chordia MD, Zigler M, Murphree LJ, Figler H, Macdonald TL, Olsson RA, Linden J (2005) 6-aryl-8H-indeno[1,2-d]thiazol-2-ylamines: A1 adenosine receptor agonist allosteric enhancers having improved potency. J Med Chem 48:5131–5139
Fawzi AB, Macdonald D, Benbow LL, Smith-Torhan A, Zhang H, Weig BC, Ho G, Tulshian D, Linder ME, Graziano MP (2001) SCH-202676: An allosteric modulator of both agonist and antagonist binding to G protein-coupled receptors. Mol Pharmacol 59:30–37
van den Nieuwendijk AMCH, Pietra D, Heitman L, Göblyös A, IJzerman AP (2004) Synthesis and biological evaluation of 2,3,5-substituted [1,2,4]thiadiazoles as allosteric modulators of adenosine receptors. J Med Chem 47:663–672
Göblyös A, de Vries H, Brussee J, IJzerman AP (2005) Synthesis and biological evaluation of a new series of 2,3,5-substituted [1,2,4]thiadiazoles as modulators of adenosine A1 receptors and their molecular mechanism of action. J Med Chem 48:1145–1151
Horstman DA, Brandon S, Wilson AL, Guyer CA, Cragoe EJ, Limbrid LE (1990) An aspartate conserved among G-protein receptors confers allosteric regulation of a2-adrenergic receptors by sodium. J Biol Chem 265:21590–21595
Garritsen A, IJzerman AP, Beukers MW, Cragoe EJ Jr, Soudijn W (1990) Interaction of amiloride and its analogues with adenosine A1 receptors in calf brain. Biochem Pharmacol 40:827–834
Garritsen A, IJzerman AP, Beukers MW, Soudijn W (1990) Chemical modification of adenosine A1 receptors. Implications for the interaction with R-PIA, DPCPX and amiloride. Biochem Pharmacol 40:835–842
Bueters TJH, Helden HPM, Danhof M, IJzerman AP (2002) Effects of the adenosine A1 receptor allosteric modulators PD 81,723 and LUF5484 on the striatal acetylcholine release. Eur J Pharmacol 454:177–182
Mizamura T, Auchampach JA, Linden J, Bruns RF, Gross GJ (1996) PD 81,723, an allosteric enhancer of the A1 adenosine receptor, lowers the threshold for ischemic preconditioning in dogs. Circ Res 79:415–423
Li X, Conklin D, Ma W, Zhu X, Eisenach JC (2002) Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain 97:117–125
Li X, Conklin D, Pan H-L, Eisenach JC (2003) Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J Pharmacol Exp Ther 305:950–955
Gao Z-G, IJzerman AP (2000) Allosteric modulation of A2A adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol 60:669–676
Gao Z-G, Melman N, Erdmann A, Kim SG, Müller CE, IJzerman AP, Jacobson KA (2003) Differential allosteric modulation by amiloride analogues of agonist and antagonist binding at A1 and A3 adenosine receptors. Biochem Pharmacol 65:525–534
Gao Z-G, van Muijlwijk-Koezen JE, Chen A, Müller CE, IJzerman AP, Jacobson KA (2001) Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol Pharmacol 60:1057–1063
van Galen PJM, Nissen P, van Wijngaargen I, IJzerman AP, Soudijn W (1991) 1H-Imidazo[4,5-c]quinolin-4-amines: Novel non-xanthine adenosine antagonists. J Med Chem 34:1202–1206
Gao Z-G, Kim S-G, Soltysiak KA, Melman N, IJzerman AP, Jacobson KA (2002) Selective allosteric enhancement of agonist binding and funtion at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol Pharmacol 62:81–89
Topliss JG (1972) Utilization of operational schemes for analog synthesis in drug design. J Med Chem 15:1006–1011
Göblyös A, Gao Z-G, Brussee J, Connestari R, Santiago SN, Ye K, IJzerman AP, Jacobson KA (2006) Structure-activity relationships of new 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric enhancers of the A3 adenosine receptor. J Med Chem 49:3354–3361
Bhattacharya S, Youkey RL, Ghartey K, Leonard M, Linden J, Tucker AL (2006) The allosteric enhancer PD 81,723 increases chimaeric A1/A2A adenosine receptor coupling with Gs. Biochem J 396:139–146
Ligt RAF, Rivkees SA, Lorenzen A, Leurs R, IJzerman AP (2005) A “locked-on,” constitutively active mutant of the adenosine A1 receptor. Eur J Pharmacol 510:1–8
Kourounakis A, Visser C, de Groote M, IJzerman AP (2001) Differential effects of the allosteric enhancer (2-amino-4,5-dimethyl-trienyl)[3-(trifluoromethyl)phenyl]-methanone (PD 81,723) on agonist and antagonist binding and function at the human wild-type and a mutant (T277A) adenosine A1 receptor. Biochem Pharmacol 61:137–144
Barbhaiya H, McClain R, IJzerman A, Rivkees SA (1996) Site-Directed mutagenesis of the human A1 adenosine receptor: Influences of acidic and hydroxy residues int he first four transmembrane domains on ligand binding. Mol Pharmacol 50:1635–1642
Gao Z-G, Jiang Q, Jacobson KA, IJzerman AP (2000) Site-directed mutagenesis studies of human A2A adenosine receptors: involvement of glu(13) and his (278) in ligand binding and sodium modulation. Biochem Pharmacol 60:661–668
Gao Z-G, Kim S-K, Gross AS, Chen A, Blaustein JB, Jacobson KA (2003) Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol Pharmacol 63:1021–1031
Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265
Acknowledgements
The authors thank Kai Ye for his expert help with Fig. 7.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Göblyös, A., IJzerman, A.P. Allosteric modulation of adenosine receptors. Purinergic Signalling 5, 51–61 (2009). https://doi.org/10.1007/s11302-008-9105-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-008-9105-3