
Abstract
Extracellular nucleotides play an important role in lung defense, but the release mechanism and relative abundance of different nucleotide species secreted by lung epithelia are not well defined. In this study, to minimize cell surface hydrolysis, we used a low-volume, flow-through chamber and examined adenosine and uridine nucleotide concentrations in perfusate aliquots of human lung A549 cells challenged by 50% hypotonic shock. Adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), and adenosine (Ado) were quantified in high-performance liquid chromatography (HPLC) analysis of fluorescent etheno derivatives, and uridine triphosphate (UTP) and uridine diphosphate (UDP) were measured using HPLC-coupled radioenzymatic assays. After the onset of hypotonic shock, ATP, ADP, UTP, and UDP in the perfusates increased markedly and peaked at approximately 2.5 min, followed by a gradual decay in the next 15–20 min; peak changes in Ado and AMP were relatively minor. The peak concentrations and fold increment (in parentheses) were: 34 ± 13 nM ATP (5.6), 11 ± 5 nM ADP (3.7), 3.3 ± 1.2 nM AMP (1.4), 23 ± 7 nM Ado (2.1), 21 nM UTP (>7), and 11 nM UDP (27). Nucleotide release was almost completely abolished from cells loaded with the calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA). Under isotonic conditions, elevation of intracellular calcium with the calcium ionophore ionomycin (5 μM, 3 min) also released nucleotides with kinetics and relative abundance as above, albeit less robust. ADP:ATP (1:3) and UDP:UTP (1:2) ratios in perfusates from stimulated cells were markedly higher than the cytosolic ratios of these species, suggesting that a nucleotide diphosphate (NDP)-rich compartment, e.g., the secretory pathway, contributed to nucleotide release. Laser confocal microscopy experiments illustrated increased FM1-43 uptake into the plasma membrane upon hypotonic shock or ionomycin treatment, consistent with enhanced vesicular exocytosis under these conditions. In summary, our results strongly suggest that calcium-dependent exocytosis is responsible, at least in most part, for adenosine and uridine nucleotide release from A549 cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Extracellular nucleotides control a diverse range of physiological processes by interacting with a large group of cell-surface P2Y and P2X receptor families [1–3]. In the lungs, extracellular nucleotides regulate airway surface liquid homeostasis and mucociliary clearance by modulating epithelial ion and fluid transport as well as ciliary beating [4]. In alveoli, adenosine triphosphate (ATP) is a potent secretagogue that stimulates type II cell-surfactant secretion. Nucleotide levels on airway surfaces, measured in vitro and ex vivo, show dynamic changes due to the combination of basal and stimulated release and their rapid metabolism by several groups of membrane-associated and membrane-secreted soluble ectoenzymes. These ectoenzymes extend the signaling potential of ATP by converting it to adenosine (Ado), a ligand of the A1–3 family of G-protein-coupled receptors [1, 4–7].
Nucleotide release is stimulated by cell mechanical perturbations, such as shear stress, membrane stretch, medium change, hyposmotic swelling, and hypoxia [1, 8]. It is now recognized that mechanosensitive ATP release occurs from healthy cells via physiological processes, which do not involve cell damage. Numerous reports have suggested that the cystic fibrosis transmembrane conductance regulator (CFTR) and other members of the superfamily of ATP-binding cassette-transport proteins serve as a conductive pathway for ATP release or regulate an associated ATP channel. However, other groups using patch-clamp, lipid-bilayer and luminometry techniques have not found any detectable CFTR-mediated or CFTR-regulated ATP release (reviewed in [1]). Volume-regulated anion channels (VRAC) and voltage-dependent anion channels (VDAC, porins or maxi-Cl− channels) are known to have considerable permeability to cations and large organic anions, and several laboratories have implicated these channels in cell-swelling-induced ATP release and the autocrine regulation of cell volume. However, recent studies have demonstrated that conductive pathways, including VRAC, VDAC, and stretch-activated channels, are not involved in cell-swelling-induced ATP release from A549 cells [1, 9, 10]. Finally, connexin hemichannels also have been proposed to mediate ATP release, but their role in this process in a physiological setting is not always clear [1].
Whereas conductive release mechanisms are still being debated, exocytotic ATP release is well established in excitatory cells, blood platelets, and chromaffin cells. Furthermore, there is growing experimental evidence supporting such a release mechanism also by epithelial and other nonexcitatory cells. In particular, we recently demonstrated that cell-swelling-induced ATP release from A549 and 16HBE14o− epithelial cells and 3T3 fibroblasts is tightly correlated with intracellular Ca2+ elevations; it is abolished in cells loaded with the calcium (Ca2+) chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) or by low temperature, strongly suggesting the involvement of Ca2+-dependent exocytosis [10]. Whether release of other nucleotides from epithelial cells also involves Ca2+-dependent exocytosis remains incompletely explored. Such studies, however, are complicated by nucleotide hydrolysis at the airway surface, which makes it difficult to assess the magnitude and relative abundance of different nucleotide species released. Therefore, we used a flow-through chamber to minimize cell surface hydrolysis.
We found that hypotonic shock markedly increased ATP, adenosine diphosphate (ADP), uridine triphosphate (UTP), and uridine diphosphate (UDP) concentrations in perfusates, which peaked at approximately 2.5 min. Nucleotide release was almost completely abolished from cells loaded with BAPTA and, under isotonic conditions, could be evoked by elevation of intracellular calcium with the calcium ionophore ionomycin. High nucleotide diphosphate (NDP) concentrations in perfusates of stimulated cells suggested that an NDP-rich compartment, e.g., the secretory pathway, contributed to this release. Together with real-time FM1-43 fluorescence experiments, our results strongly indicate that calcium-dependent exocytosis is a major mechanism of adenosine and uridine nucleotide release from A549 cells.
Materials and methods
Cells
Human lung carcinoma A549 cells were grown in Dulbecco’s modified eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 56 U/ml penicillin-G and 56 μg/ml streptomycin sulfate. All constituents of the culture media were from GIBCO-BRL (Burlington, ON, Canada). ATP efflux was measured from cell monolayers grown to confluency on 24 × 60-mm glass coverslips. Fura-2 calcium imaging and FM1-43 microscopy experiments were performed on cells grown on circular 15-mm diameter no. 1 glass coverslips.
Nucleotide efflux assay
To measure nucleotide efflux during hypotonic challenge with high temporal resolution, we used a custom-designed, low-volume (300 μl), flow-through chamber, as described previously [10]. Briefly, 24 × 60-mm glass coverslips with confluent cell monolayers of ∼500 cells/mm2 were mounted in the chamber and perfused with warm (37°C/in-line heater; Warner Instrument, Hamden, CT, USA) Ringer solution at the rate of 1.3 ml/min. The Ringer solution contained (in mM): 110.5 NaCl, 24 NaHCO3, 1.3 KCl, 1 MgCl2, 1 CaCl2, 2.5 Na2HPO4, 2.5 KH2PO4, 1.2 K2HPO4, and 10 glucose; pH 7.4 was maintained by bubbling with 5% CO2. After an equilibration period in isotonic solution (5–15 min), a 50% hypotonic solution was applied, and the perfusate was continuously collected for 30-s intervals during the initial burst of ATP secretion (0–5 min) and during 1 min elsewhere. Nucleotide concentrations in the samples were evaluated by high-performance liquid chromatography (HPLC0 analysis, as described below. For studies in absence of extracellular calcium, CaCl2 was omitted, and the solutions were supplemented with 0.1 mM ethyleneglycoltetraacetic acid (EGTA). The 50% hypotonic solution was prepared by appropriate reduction of salt concentration, whereas divalent cation concentrations were kept constant.
HPLC quantification of adenine and uridine nucleotides
UTP concentrations were quantified by UDP-glucose pyrophosphorylase-based reaction [11]. Briefly, 100-μl samples were incubated in the presence of 0.5 U/ml UDPglucose pyrophosphorylase, 0.5 U/ml inorganic pyrophosphatase, 1.6 mM CaCl2, 2 mM MgCl2, 25 mM hydroxyethylpiperazine ethanesulfonic acid (HEPES) (pH 7.4), and ∼100,000 cpm 1 μM [14C]glucose-1P. Incubations lasted 1 h at 30°C. Reactions were terminated by heating the samples at 95°C for 2 min. Conversion of [14C]glucose-1P to [14C]UTP was determined by HPLC (Shimadzu) via a Nova Pack C18 column and ion-pairing mobile phase. Radioactivity was measured on-line with a Packard Flo-One detector. Assay sensitivity was 1 pmol (3 nM in 100-μl samples).
UDP was quantitatively phosphorylated in the presence of [γ32P]ATP using nucleoside diphosphokinase (NDPK). Briefly, 100-μl samples containing 0.1 U/ml NDPK, 0.1 μCi 60 nM [γ32P]ATP, 1.6 mM CaCl2, 2 mM MgCl2, and 25 mM HEPES (pH 7.4) were incubated for 5 min at 30°C. Reactions were terminated by heating the samples at 95°C for 2 min. The resulting conversion of [γ32P]ATP to [γ32P]UTP was monitored by HPLC [12]. A calibration curve employing known amounts of UDP was chartered in parallel during each assay. This assay allows UDP quantification with sensitivity of 20 fmol (0.2 nM in 100-μl samples).
For etheno (ɛ) derivatization, samples (200 μl) were derivatized for 30 min at 72°C in the presence of 1.0 M chloroacetaldehyde and 25 mM Na2HPO4 (pH 4.0). The resulting fluorescent 1,N6-ethenoadenine derivatives were analyzed by HLPC (Waters) in a Hamilton PRP-×100 anion exchange column, as described previously [4]. ɛ-ATP, ɛ-ADP, ɛ-AMP, and ɛ-Ado were quantified with a sensitivity of 200 fmol (1 nM in 200-μl samples).
FM1-43 studies
Bulk exocytosis was quantified in A549 cells as changes in the fluorescence intensity of FM1-43 incorporated into the plasma membrane [13, 14] and recorded by real-time confocal microscopy. Cells were washed with Hank’s balanced salt solution + 20 mM HEPES + 2 mM MgCl2 and 1.6 mM CaCl2 (HBSSH) or with HBSSH devoid of calcium (HBSSH 0 Ca). They were mounted onto the stage of a Leica SP2 AOBS confocal microscope equipped with HCX Apo L63x NA 0.9 immersion Leica lens and a 488-nm Argon laser. Experiments were initiated by incubation of the cells with 3 μM FM1-43. Cells were incubated for at least 10 min, and hypotonic shock was applied by decreasing salt concentration 33% while maintaining calcium, magnesium, and FM1-43 concentrations constant. Alternatively, cells incubated with FM1-43 in HBSSH or HBSSH 0 Ca were stimulated with 5- to 10-μM ionomycin. Real-time recording was performed by laser scanning in thexz axes with a galvostage, initially every 10 s and then every 30 s for the time periods indicated in the figures. Overall fluorescence intensity changes associated with the plasma membrane were estimated by measuring the intensity value associated with each pixel through time. The entire apical membrane compartment displayed in a confocal plane and five random regions of basolateral and subapical domains were analyzed, normalized to basal values (time = 0), and averaged for each region. Cell swelling was estimated as a change of cell height in thexz plane at different time points, normalized to basal values, and averaged.
Fura-2 calcium measurements
To load Fura-2, cells were incubated (1 h, 37°C, 5% CO2) in physiological solution containing 25 μM Fura-2-AM + 0.02% Pluronic F127 and 2.5 mM probenecid. This was followed by 30 min deesterification period in physiological solution containing probenecid. The physiological saline solution consisted of (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 glucose and 10 HEPES, pH 7.4, adjusted with sodium hydroxide (NaOH). Fifty percent hypotonic medium was prepared by reducing the salt concentration while keeping divalent cation concentration constant. For calcium imaging, coverslips with Fura-2-loaded cells were mounted in the imaging/perfusion chamber attached to the heated platform (Warner Instruments) on the stage of an inverted microscope (Nikon TE300). The cells were exposed to alternate (200 ms) illumination at 340 nm and 380 nm with a high-pressure mercury lamp (100 W) via interference filters (Chroma Technology, Brattleboro, VT, USA) mounted on a filter wheel (Sutter Lambda 10-C, Sutter Instrument, Novato, CA, USA) and a dichroic mirror (510/540 nm, Chroma Technology). Fluorescence images were recorded at 15- to 60-s intervals with the digital camera and stored for later analysis.
Chemicals
For calcium-imaging experiments, Fura-2-AM was obtained from Molecular Probes, Invitrogen Corp. (Burlington, ON, Canada). Probenicid, Pluronic F127 and all other reagents were from Sigma Aldrich (Oakville, ON, Canada).
Results
Kinetics of nucleotide release
Figure 1 shows an example of the time course of nucleotide release induced by 50% hypotonic shock. For clarity, the release of adenine and uridine nucleotides appears on separate graphs: a and b, respectively. The kinetics of release were remarkably similar for all nucleotides. After the onset of hypotonic shock, nucleotide concentration increased rapidly, peaking at ∼2.5 min, followed by a gradual decay in the next 10–15 min. The average peak values from several separate experiments are shown in Fig. 1c. Interestingly, ATP was the major species at the peak of stimulated release, whereas for basal release, Ado was the predominant species. The rank order of nucleotide abundance at the peak was: ATP > Ado ≥ UTP > ADP ≈ UDP > AMP. Relative increases of nucleotide conconcentrations at the peak were also the highest for nucleotide triphosphates (NTPs) and nucleotide diphosphates (NDPs) (ATP 5.6-fold, ADP 3.7-fold, UTP >7-fold, UDP 27-fold), whereas the increase was smaller for AMP and Ado (1.4-fold and 2.1-fold, respectively).

Transient nucleotide release from A549 cells induced by 50% hypotonic shock. Time course of adenosine (a) and uridine (b) nucleotide release observed in response to 50% hypotonic shock. A representative experiment is shown out of four performed under the same conditions. Hypotonic shock was applied at t = 0 min and was preceded by 15-min equilibration in isotonic solution. Basal (t = 0 min) and peak (t = 2.5 min) nucleotide concentrations detected in perfusates (c). Average values [± standard deviation (SD)] are from three to four experiments, such as in a and b, except for uridine triphosphate (UTP) and uridine diphosphate (UDP), which are from a single experiment. * Due to the limited sensitivity of UTP evaluations (≈3 nM), the basal level of UTP was found to be below the detection limit and was not shown
Role of intracellular calcium ([Ca+2]i)
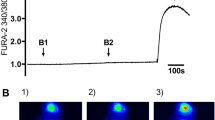
To investigate the role of intracellular calcium ([Ca+2]i) in adenine nucleotide release, we tested the effect of the Ca2+ ionophore ionomycin. In the absence of hypotonic shock, exposure to 5 μM ionomycin for 3 min induced transient nucleotide release from A549 cells (Fig. 2a). This release reached a peak at approximately 1.5 min, i.e., slightly earlier and at somewhat lesser absolute peak amplitude compared with that induced by 50% hypotonic shock. Otherwise, the kinetics and relative nucleotide abundance were similar for both stimuli. Almost complete inhibition of swelling-induced adenine nucleotide release was observed for A549 cells loaded with the Ca2+ chelator BAPTA, compared with control untreated cells of the same batch tested in parallel experiments (Fig. 2b, c, respectively). Fura-2 fluorescence [Ca+2]i measurements confirmed a dramatic reduction of the [Ca+2]i response to 50% hypotonic shock in BAPTA-loaded cells (Fig. 2d). These experiments demonstrate that elevation of [Ca+2]i was required to trigger adenine nucleotide release from A549 epithelial cells.

Effect of intracellular calcium ([Ca2+ i]) modulators on nucleotide release. (a). In the absence of hypotonic shock, application of 5 μM ionomycin resulted in transient nucleotide release with kinetics similar to that induced by hypotonic shock. An example, out of two similar experiments, is shown. Loading A549 cells with the Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) almost completely inhibited hypotonic-stress-induced nucleotide release compared with controls that were run in parallel (b and c, respectively) (n = 2). Effect of BAPTA on the [Ca2+]i response (d). The two traces represent changes of the Fura-2 fluorescence ratio at two excitation wavelengths, λ340/λ380, in response to 50% hypotonic shock, applied at t = 0 min, in control and BAPTA-loaded cells. Note that the rapid peak of the [Ca2+]i response was abolished in BAPTA-loaded cells. Similar responses were observed in four experiments
FM1-43 fluorescence changes implicate vesicular exocytosis
Strong calcium dependence of nucleotide release may indicate the involvement of vesicular exocytosis. To further explore this possibility, bulk exocytosis was examined by real-time confocal microscopy in cells bathed in FM1-43. Fluorescence intensity associated with the plasma membrane increased rapidly by incorporation of the soluble probe into the membrane and remained almost constant after 5 min in nonstimulated cells. Plasma-membrane-associated fluorescence intensity rose rapidly (∼20 s) with hypotonic shock stimulation. This change was accompanied by increased cell volume seen as increment of cell height (Fig. 3a, b). Cell volume peaked at about 3 min, followed by a regulatory volume decrease in the next 10–15 min (data not shown). A similar plasma-membrane-associated FM1-43 fluorescence intensity increase was observed in the absence of hypotonic shock when the cells were stimulated with ionomycin in the presence but not in the absence of calcium in the bathing solution (Fig. 4). These data indicate that both hypotonic-shock- and ionomycin-stimulated bulk exocytosis in A549 cells.

Effect of hypotonic shock on FM1-43 surface membrane staining. Confocal FM1-43 fluorescence images of confluent A549 cells after 20 min of dye loading in isotonic solution, followed by 50% hypotonic shock stimulation (a). The images depict the xz scans of the cell monolayer at different time points. Note the increase in cell size and fluorescence intensity consistent with hypotonic-shock-induced cell swelling and heightened bulk exocytosis. Time course of FM1-43 plasma membrane fluorescence and cell height changes during hypotonic shock (b)

Effect of ionomycin on FM1-43 surface-membrane staining. Confocal FM1-43 fluorescence images of confluent A549 cells prior to and after 4-min exposure to 5 μM ionomycin in the presence of extracellular calcium. (a). The images show the xz scans of the cell monolayer at different time points. Note the increase in fluorescence intensity after ionomycin stimulation, indicating enhanced exocytosis. Time course of FM1-43 plasma membrane fluorescence changes of unstimulated cells (Δ CON) and cells during ionomycin treatment in the presence (▪ ION) or absence (• ION 0 Ca) of extracellular calcium (b)
Discussion
In this study, we used the flow-through chamber and an etheno derivatization measurement technique to examine the kinetics of nucleotide release from A549 lung epithelial cells induced by hypotonic shock. We demonstrated that ATP, ADP, AMP, Ado, UTP, and UDP appeared in the perfusates with the same kinetics, peaking at ∼2.5 min (Fig. 1), which coincided with the peak of [Ca+2]i elevation evoked by hypotonic shock (Fig. 2d). Similar tight temporal correlation between the [Ca+2]i signal and ATP release, measured by luciferase-luciferin luminescence assay, was reported previously for A549, 16HBE14o− epithelial cells and NIH3T3 fibroblasts [10]. Such a close temporal association suggests that hypotonic-shock-induced elevation of [Ca+2]i is a trigger for the release of nucleotides. This was supported by the strong inhibitory effect of BAPTA-AM and the induction of nucleotide release in the absence of hypotonic shock solely by the rise of [Ca+2]i with ionomycin. These data demonstrate tight Ca2+ dependence of nucleotide release, and point to a Ca2+-dependent exocytotic mechanism. It should be noted that the bulk of [Ca+2]i changes shown in Fig. 2d likely represent superposition of spatially separated, distinct [Ca+2]i responses, one evoked directly by hypotonic shock and the other due to autocrine/paracrine actions of the released nucleotides on P2Y receptors. The latter effects might be minimized, at least partially, by continuous perfusion, which reduced nucleotide concentration in the bulk of the chamber far below the IC50 of P2Y receptor activation; however, this possibility requires further study.
Consistent with a Ca2+-dependent exocytotic mechanism, FM1-43 fluorescence studies revealed enhanced incorporation of the probe into the plasma membrane induced by hypotonic shock or ionomycin treatment. Increased plasma membrane-associated FM1-43 fluorescence intensity was a measure of the cumulative amount of membrane added by exocytosis [13]. FM1-43 fluorescence increased rapidly during the first 20 s of hypotonic shock (Fig. 3). Interestingly, the initial fluorescence increment exceeded the cell-surface increase estimated from xz confocal scans. In our previous study using a dual-image 3D cell reconstruction technique [15], we observed only an 11–30% surface increase of single substrate-attached A549 cells swollen in 50% hypotonic solution [10]. This difference could be attributed in part to different methods of cell-height and surface evaluation in that study, as well as differences in swelling responses of single isolated cells compared with confluent cell monolayer. However, a similar discrepancy between FM1-43 fluorescence, cell-surface and membrane electrical capacitance has been also reported by others, e.g., with pituitary lactotrophs, where the dense granules docked into the plasma membrane were intensely stained by FM1-43 in addition to the membrane added to the cell surface [16]. Therefore, the divergence of fluorescence and the cell-surface changes seen in our study with A549 cells may also result in part from FM1-43 staining of the lipophilic content of fused vesicles in addition to the staining of fused vesicular membranes. The FM1-43 dye is often employed to fluorescently marked surfactant-containing lamellar bodies in alveolar type II (ATII) cells [17, 18] and A549 cells, a model of ATII cells, also contain granules enriched in surface-active phospholipids [19]. However, the exact contribution of such granules to increased FM1-43 fluorescence in stimulated A549 cells was out of the scope of the present investigation and will require future direct study.
Vesicle exocytosis may be mechanistically associated with nucleotide release in two ways: nucleotides can be delivered to the extracellular medium as cargo molecules within exocytotic vesicles or, alternatively, secreted from the cytosol via vesicle-associated nucleotide-conducting channels or transporters transiently expressed at the plasma membrane as a consequence of vesicle-plasma membrane fusion. Analysis of the relative abundance of different nucleotide species that appear in the extracellular medium may help to distinguish between these two mechanisms. ATP and UDP sugars accumulate in the lumen of the secretory pathway up to 20- to 50-fold above their cytosolic concentrations, and luminal utilization of these molecules generates ADP and UDP. UDP is the major uridine nucleotide detected in ER/Golgi fractions [20] and, eventually, a UDP-selective apyrase (UDPase) converts UDP to urodine monophosphate (UMP) [21]. ER/Golgi ADP and UMP are exchanged for cytosolic ATP and UDP glucose via specific transporters [22–25]. As intraluminal nucleotides are not subject to the mechanisms that retrieve resident ER/Golgi proteins, they are predicted to remain within trafficking vesicles and to be released to the extracellular space from the secretory pathway. Our results suggest that this mechanism likely accounted for UDP release from A549 cells. Figure 1c illustrates that hypotonic stimulation promoted a sharp increase in UDP levels, with a UTP:UDP concentration ratio of 2:1. As the cytosolic UTP:UDP ratio is >10:1 [26], our results suggest, at least in part, a vesicular rather than a cytosolic source of UDP and, as a corollary, of ATP and ADP.
In summary, our results provide strong evidence for Ca2+-dependent vesicular exocytosis as a major mechanism of adenosine and uridine nucleotide release from A549 epithelial cells induced by hypotonic stress. Part of this release involves vesicles of the protein secretory pathway. Further investigations are needed to clarify the origin and contribution of other vesicular pools as well as the mechanisms and sources of intracellular Ca2+ elevations that evoke nucleotide secretion.
References
Lazarowski ER, Boucher RC, Harden TK (2003) Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol Pharmacol 64:785–795
Burnstock G (2006) Historical review: ATP as a neurotransmitter. Trends Pharmacol Sci 27:166–176
Burnstock G (2006) Purinergic signalling. Br J Pharmacol 147(Suppl 1):S172–S181
Lazarowski ER, Tarran R, Grubb BR, Van Heusden CA, Okada S, Boucher RC (2004) Nucleotide release provides a mechanism for airway surface liquid homeostasis. J Biol Chem 279:36885–36864
Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 362:299–309
Donaldson SH, Lazarowski ER, Picher M, Knowles MR, Stutts MJ, Boucher RC (2000) Basal nucleotide levels, release, and metabolism in normal and cystic fibrosis airways. Mol Med 6:969–982
Boucher RC (2003) Regulation of airway surface liquid volume by human airway epithelia. Pflugers Arch 445:495–498
Grygorczyk R, Hanrahan JW (1997) CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. Am J Physiol 272:C1058–C1066
Boudreault F, Grygorczyk R (2002) Cell swelling-induced ATP release and gadolinium-sensitive channels. Am J Physiol Cell Physiol 282:C219–C226
Boudreault F, Grygorczyk R (2004) Cell swelling-induced ATP release is tightly dependent on intracellular calcium elevations. J Physiol 561:499–513
Lazarowski ER, Harden TK (1999) Quantitation of extracellular UTP using a sensitive enzymatic assay. Br J Pharmacol 127:1272–1278
Lazarowski ER, Boucher RC, Harden TK (2000) Constitutive release of ATP and evidence for major contribution of ecto- nucleotide pyrophosphatase and nucleoside diphosphokinase to extracellular nucleotide concentrations. J Biol Chem 275:31061–31068
Smith CB, Betz WJ (1996) Simultaneous independent measurement of endocytosis and exocytosis. Nature 380:531–534
Cochilla AJ, Angleson JK, Betz WJ (1999) Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci 22:1–10
Boudreault F, Grygorczyk R (2004) Evaluation of rapid volume changes of substrate-adherent cells by conventional microscopy 3D imaging. J Microsc 215:302–312
Brumback AC, Lieber JL, Angleson JK, Betz WJ (2004) Using FM1-43 to study neuropeptide granule dynamics and exocytosis. Methods 33:287–294
Ashino Y, Ying X, Dobbs LG, Bhattacharya J (2000) [Ca(2+)](i) oscillations regulate type II cell exocytosis in the pulmonary alveolus. Am J Physiol Lung Cell Mol Physiol 279:L5–L13
Mair N, Haller T, Dietl P (1999) Exocytosis in alveolar type II cells revealed by cell capacitance and fluorescence measurements [In Process Citation]. Am J Physiol 276:L376–L382
Shapiro DL, Nardone LL, Rooney SA, Motoyama EK, Munoz JL (1978) Phospholipid biosynthesis and secretion by a cell line (A549) which resembles type II aleveolar epithelial cells. Biochim Biophys Acta 530:197–207
Fleischer B (1981) The nucleotide content of rat liver Golgi vesicles. Arch Biochem Biophys 212:602–610
Wang TF, Guidotti G (1998) Golgi localization and functional expression of human uridine diphosphatase. J Biol Chem 273:11392–11399
Wang TF, Guidotti G (1998) Golgi localization and functional expression of human uridine diphosphatase. J Biol Chem 273:11392–11399
Hirschberg CB, Robbins PW, Abeijon C (1998) Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu Rev Biochem 67:49–69
Waldman BC, Rudnick G (1990) UDP-GlcNAc transport across the Golgi membrane: electroneutral exchange for dianionic UMP. Biochemistry 29:44–52
Ishida N, Kawakita M (2004) Molecular physiology and pathology of the nucleotide sugar transporter family (SLC35). Pflugers Arch 447:768–775
Kochanowski N, Blanchard F, Cacan R, Chirat F, Guedon E, Marc A, Goergen JL (2006) Intracellular nucleotide and nucleotide sugar contents of cultured CHO cells determined by a fast, sensitive, and high-resolution ion-pair RP-HPLC. Anal Biochem 348:243–251
Acknowledgements
This study was supported in part by the Canadian Institutes of Health Research and the Canadian Cystic Fibrosis Foundation (CCFF) (to RG), and by National Institutes of Health (NIH) R01 HL076303 (to EL). ST was the recipient of a CCFF studentship. The authors acknowledge the editorial assistance of Ovid Da Silva, Research Support Office, Research Centre, CHUM.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Tatur, S., Kreda, S., Lazarowski, E. et al. Calcium-dependent release of adenosine and uridine nucleotides from A549 cells. Purinergic Signalling 4, 139–146 (2008). https://doi.org/10.1007/s11302-007-9059-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-007-9059-x