
Abstract
There is abundant evidence that extracellular ATP and other nucleotides have an important role in pain signaling at both the periphery and in the CNS. The focus of attention now is on the possibility that endogenous ATP and its receptor system might be activated in chronic pathological pain states, particularly in neuropathic and inflammatory pain. Neuropathic pain is often a consequence of nerve injury through surgery, bone compression, diabetes or infection. This type of pain can be so severe that even light touching can be intensely painful; unfortunately, this state is generally resistant to currently available treatments. In this review, we summarize the role of ATP receptors, particularly the P2X4, P2X3 and P2X7 receptors, in neuropathic and inflammatory pain. The expression of P2X4 receptors in the spinal cord is enhanced in spinal microglia after peripheral nerve injury, and blocking pharmacologically and suppressing molecularly P2X4 receptors produce a reduction of the neuropathic pain behaviour. Understanding the key roles of these ATP receptors may lead to new strategies for the management of intractable chronic pain.
Similar content being viewed by others


Introduction
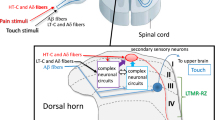
Extracellular ATP might be an important substrate in the formation of pain [1–7]. The first clue to this possibility was found about 30 years ago in clinical studies showing that ATP applied to blister bases [1, 2] induced a pain sensation in humans. Significant advances in our understanding of mechanisms by which ATP causes pain have been made recently by the discovery of cell-surface receptors for detecting extracellular ATP and other nucleotides on sensory neurons [3–7]. In a subset of primary afferent sensory neurons, ATP or its analogues produce electrophysiological and biological responses via ligand-gated ion-channel receptors, namely P2X receptors (P2XRs) [8–13], and G protein-coupled receptors, namely P2Y receptors (P2YRs) [14–19]. However, blocking P2XRs or P2YRs pharmacologically or suppressing their expression in sensory neurons or spinal cord had little effect on acute physiological pain evoked by heat or mechanical pressure in normal animals [20–22], although inflammatory pain was attenuated [20, 23]. It seems likely that endogenous ATP and its receptor system may be more prominent in chronic pain states, particularly in neuropathic pain or inflammatory pain, than in normal conditions [22–29].
Neuropathic pain, which often develops when nerves are damaged through surgery, cancer, bone compression, diabetes or infection, is a type of pathological pain that does not resolve even when the overt tissue damage has healed [30–32]. Neuropathic pain is so severe that even light contact with clothing can cause intense pain (tactile allodynia: an abnormal hypersensitivity to innocuous stimuli) and is often resistant to most current treatments, including morphine. Accumulating evidence concerning how peripheral nerve injury causes neuropathic pain has suggested that molecular and cellular alterations in the neuronal circuit between primary sensory neurons and spinal dorsal horn neurons after nerve injury have an important role in the pain [30–32]. Several reports suggest that P2X3Rs [25–27, 33, 34] or P2X7 [35, 36] have a role in neuropathic pain. And, we recently revealed that the P2X4R subtype in the spinal microglia is required for the expression of neuropathic pain [37]. More recently, we have reported that BDNF released from microglia by the stimulation of P2X4 causes the shift in neuronal anion gradient underlying neuropathic pain [38]. Here we review the progress in the current understanding of how these ATP receptors act in the pathophysiology of neuropathic and inflammatory pain.
Microglial P2X4 and nerve injury-induced pain
Glial cells make up over 70% of the total cell population in the CNS and are classified into astrocytes, oligodendrocytes and microglia. Microglia are ubiquitously distributed in the CNS and represent a morphologically unique type of cell. In normal conditions, microglia are referred to as ‘resting microglia–and have a small soma bearing thin and branched processes [39, 40]. Once activated by neuronal injury, trauma, ischemia, infection, or neurological diseases, microglia are referred to as ‘activated microglia–and show a stereotypic changes in morphology (amoeboid shape), gene expression, function and number [39–41]. The changes in expression of cell-surface molecules (i.e., complement receptor 3, which is recognized by the antibody OX42) and in morphology are widely used as the key diagnostic markers of activated microglia [39, 40].
Clinical evidence that neuropathic pain results from damage to peripheral nerves in humans led to the development of a variety of animal models for studying neuropathic pain. In most animal models of neuropathic pain which have been intensively studied peripheral nerves are directly damaged [42–46]. In such models, a dramatic change in microglia within the spinal dorsal horn has been reported after the nerve injury [47–51]. Within the first 24 h after peripheral nerve injury, spinal microglia become hypertrophic in their short and thick processes [47]. This is followed by a burst proliferation of microglia with a peak at around 2– days after the nerve injury [52]. Activated microglia exhibit upregulated OX42 labeling in the dorsal horn [47, 49–53], which starts to increase as early as 1 day after nerve injury and peaks at around 14 days [49]. The pattern of OX42 upregulation in the dorsal horn correlated with that of the development of tactile allodynia [49]. Although there have been many studies showing that activation of microglia in the dorsal horn is correlated with the development of pain hypersensitivity in a wide variety of nerve injury models [47, 49–54], it remained an open question whether spinal microglia play a causal role in neuropathic pain behaviour.
A clue to identifying P2X4 in the spinal cord as being required for neuropathic pain first came from pharmacological investigations using the P2X antagonists TNP-ATP and PPADS [37]. We found that the marked tactile allodynia that develops following the nerve injury was reversed by acutely administering TNP-ATP intrathecally but was unaffected by administering PPADS. TNP-ATP had no effect on acute pain behaviour in the uninjured state or on motor behaviour. From the pharmacological profiles of TNP-ATP (blocking P2X4 at high concentration) and PPADS (not blocking P2X4), it was inferred that tactile allodynia depends upon P2X4 in the spinal cord. The expression of P2X4 protein, normally low in the naive spinal cord, progressively increased in the days following nerve injury with a time-course parallel to that of the development of tactile allodynia. Double immunolableling analysis using cell-specific markers demonstrated that microglia in the dorsal horn on the side of the nerve injury were intensely positive for P2X4 protein. The cells expressing P2X4 in the nerve-injured side of the dorsal horn were more numerous than under control conditions and showed high levels of OX42 labeling and morphological hypertrophy, all of which are characteristic markers of activated microglia. Moreover, it was found that reducing the expression of P2X4 protein in spinal microglia by means of intrathecally administered antisense oligodeoxynucleotide targeting P2X4R prevented the development of the nerve injury-induced tactile allodynia. Collectively, this evidence implies that activation of microglial P2X4 is necessary for pain hypersensitivity following nerve injury.
The sufficiency of P2X4 activation in microglia for the development of allodynia was demonstrated by intrathecal administration of activated microglia stimulated in vitro by ATP [37]. In otherwise naive animals, intrathecal administration of cultured microglia that were preincubated with ATP to activate P2X4 on microglia produced tactile allodynia progressively over the 3– h following the administration. Microglia also express another subtype of P2X, P2X7, but this receptor subtype appears not to be involved because activation of P2X7 typically requires a higher concentration of ATP than used [55, 56] and because TNP-ATP, which does not affect P2X7 [57], prevents ATP from stimulating microglia to produce allodynia. These findings indicate that P2X4 stimulation of microglia is not only necessary for tactile allodynia, but is also sufficient to cause the allodynia.
Possible mechanisms underlying microglial P2X4-related neuropathic pain
The variety of biological effects produced by ATP in microglia during in vitro studies may provide hints towards clarifying the mechanisms by which microglia produce altered processing of information in the spinal cord dorsal horn. It was recently shown that ATP-stimulated microglia signal to lamina I neurons, causing a collapse of their transmembrane anion gradient, and that brain-derived neurotrophic factor (BDNF) is a crucial signaling molecule between microglia and neurons [38]. Since it was already reported that the nerve injury-induced tactile allodynia depends on a depolarizing shift in the Eanion of spinal lamina I (LI) neurons in the dorsal spinal cord, resulting in converting the GABAA-receptor- and glycine-receptor-mediated inhibition to excitation [58], it was considered that microglia may affect Eanion in LI neurons. To investigate this possibility, microglia were administered to the lumbar spinal level of naive rats by an intrathecal catheter as described [37]. Administering microglia stimulated with ATP caused a progressive tactile allodynia over the 5 h after injection. Voltage-clamp recording were made from LI neurons of slices prepared 5 h after intrathecal microglia administration. Eanion in LI neurons from rats administered ATP-stimulated microglia was shifted to −?1.6 mV from −?8.3 mV that is Eanion in spinal slices taken from normal rats. In addition, using current clamp recordings, GABA response switched from hyperpolarizing in control rats to depolarizing in rats treated with ATP-stimulated microglia. Activated microglia secrete various biologically active molecules, one of which, BDNF, was implicated in the hypersensitivity of dorsal horn neurons that follows sensitization and inflammation [59–61] and in anion gradient shifts in the hippocampus [62].
To examine whether BDNF could trigger shifts in pain hypersensitivity and in LI neuronal Eanion similar to those resulting from the application of ATP-stimulated microglia, recombinant BDNF was administered intrathecally to normal rats. BDNF produced tactile allodynia comparable to that produced by ATP-stimulated microglia. Eanion of LI neurons in slices treated with BDNF (>90 min, in vitro) was significantly less negative than that of LI neurons from control slices. During perfusion with BDNF and in the presence of glutamate receptor blockers, the proportion of neurons responding to GABA with a rise in [Ca2+]i increased over time, reaching 31% of neurons recorded between 80 and 120 min. The rise in [Ca2+]i was prevented by the GABAA receptor blocker bicuculline, confirming that the effect was mediated by GABAA receptors. Thus, acute administration of BDNF in slices caused a depolarizing shift in Eanion and caused GABA to produce net excitation. To examine the effects of prolonged exposure to BDNF in vivo, a BDNF-transducing recombinant adenovirus (adBDNF) [63] was administered intrathecally to the rats. A progressive tactile allodynia was observed over the 4 days after the treatment of adBDNF. Eanion in LI neurons from adBDNF-injected rats was significantly less negative than that in LI neurons from control rats. Thus, similar to acute administration of BDNF, sustained local release of BDNF caused the allodynia and a depolarizing shift in Eanion. Moreover, a function-blocking antibody against the TrkB receptor (anti-TrkB) and a BDNF-sequestering fusion protein (TrkB-Fc) acutely inhibited the allodynia and the shift of Eanion of LI neurons. These findings indicate that endogenous BDNF is necessary to sustain both the tactile allodynia and the depolarizing shift in Eanion in LI neurons that result from nerve injury.
The administration of ATP-stimulated microglia with either anti-TrkB or TrkB-Fc did not develop tactile allodynia. After pretreatment of microglia with double-stranded short interfering RNA directed against BDNF (BDNF siRNA), the ATP-stimulated microglia injected intrathecally into normal rats did not cause the allodynia. Anti-TrkB and BDNF siRNA prevented the shift in Eanion induced by ATP-stimulated microglia. ATP stimulation caused release of BDNF from microglia in culture. This effect of ATP was blocked by treating the cultures with the P2X receptor blocker TNP-ATP. In addition, pretreatment of the microglia with BDNF siRNA prevented release of BDNF by ATP stimulation. By bath-application of TNP-ATP to spinal slices taken from allodynic rats 2 weeks after nerve injury, Eanion of LI neurons was returned to normal value. Thus, P2X4 receptor activation is necessary to sustain the depolarizing shift in Eanion in rats with nerve injury. These findings show that both the decrease in paw withdrawal threshold and the shift in Eanion in LI neurons caused by ATP-stimulated microglia through P2X4 require BDNF-TrkB signaling and that the source of BDNF is the microglia themselves.
Possible involvement of P2X7 in chronic pain: TNF-α
Several cytokines, such as IL-1β, IL-6 and TNF-α, in the dorsal horn are increased after nerve lesion [64–66] and have been implicated in contributing to neuropathic pain [54, 64–68].
Recent evidence indicates the relationship between TNF-α and neuropathic pain [69–73] and TNF-α released after injury is proposed as an initiator of abnormal pain sensation [69–72]. TNF-α is upregulated after nerve injury in both the DRG [73, 74] and spinal cord [64–66, 75]. The inhibition of TNF-α reduces the hyperalgesia in neuropathic pain models [76, 77]. After peripheral nerve injury, DRG neurons robustly increase their expression of TNF-α [73]. Exogenous TNF-α applied to intact or compression-injured DRG induces sustained mechanical allodynia [78]. However, the mechanisms by which TNF-α elicits pain behavior are still unclear. Previous studies suggest that TNF-α modulates neuronal activity in neurons [79–85]. Schäfers et al. [74] investigated responses of intact and nerve-injured DRG neurons to locally applied TNF-α using parallel in vivo and in vitro paradigms. In vivo, TNF-α (0.1–0 pg/ml) or vehicle was injected into L5 DRG in naive rats and in rats that had received L5 and L6 spinal nerve ligation (SNL) immediately before injection. In naive rats, TNF-α elicits long-lasting allodynia. In SNL rats, subthreshold doses of TNF-α synergize with nerve injury to elicit faster onset of allodynia and spontaneous pain behavior. Pre-emptive treatment with etanercept, a TNF-α antagonist, reduces SNL-induced allodynia by almost 50%. Perfusion of TNF-α (100–,000 pg/ml) to naive DRG neuron evokes short lasting discharges. In injured DRG, TNF-α elicits higher and longer lasting neuronal discharges in earlier onset at much lower concentrations. In naive DRG which is adjacent to injured DRG, TNF-α also elicits high-frequency discharges at subthreshold concentrations. These data suggest that injured and adjacent uninjured DRG neurons are sensitized to TNF-α after SNL, and sensitization to endogenous TNF-α may be essential for the development and maintenance of neuropathic pain.
Microglia are a major source of TNF-α. TNF-α following an increase in the TNF-α mRNA expression by activating MAPKs through P2X7 in microglia. ATP potently stimulates the release of TNF-α following an increase in the TNF-α mRNA expression by activating MAPKs [86, 87]. The TNF-α release is maximally elicited by 1 mM ATP and also induced by a P2X7 receptor agonist, BzATP, suggesting the involvement of P2X7 receptor [86]. ATP-induced TNF-α release is Ca2+-dependent, and a sustained Ca2+ influx correlates with the TNF-α release. ATP-induced TNF-α release needs MAPKs activation. U0126, SP600125 and SB 203580, which inhibit MEK (MAPK kinase), JNK (c-Jun N-terminal kinase), and p38, respectively, all potently suppress the production of TNF-α protein in ATP-stimulated microglia, whereas the production of TNF-α mRNA is strongly inhibited by U0126 and SP600125 but not by SB203580. This suggests that a transcription of TNF-α mRNA is dependent on both of ERK and JNK, but not on p38. SB203580 does not affect the increased levels of TNF-α mRNA but does prevent TNF-α mRNA from accumulating in the cytoplasm, suggesting that p38 plays an important role in the nucleocytoplasmic transport of TNF-α mRNA. The ATP-induced activation of JNK and p38, but not ERK are inhibited by brilliant blue G, a P2X7 receptor blocker, and by genistein and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-D] pyrimidine, which are general and src-family-specific tyrosine kinase inhibitors, respectively. These findings indicate that an src family acts downstream of the P2X7 receptor to activate JNK and p38 independently from channel action [87].
In cultured DRG neurons, exogenous TNF-α activates p38 MAPK [88]. Recently, p38 activation is shown to play a major role in the maintenance of pain [89–92]. It is speculated that activation of the p38 cascade may represent a route correlating the development of pain after nerve injury. To obtain the answer the question, it is investigated whether TNF-α activates the p38 cascade in vivo to trigger pain behavior after SNL [93]. As the result, etanercept treatment starting 2 days before SNL attenuates mechanical allodynia. Interestingly, the treatment starting 1 or 7 days after SNL is ineffective. Similarly, intrathecal infusion of a p38 inhibitor (SB203580, 4 mg/day) is effective only when it is started before but not 7 days after SNL. In DRG, activated p38 is transiently elevated 5 h after SNL and returns to baseline by 1 day after SNL. Phosphorylated p38 is localized in small TNF-α-positive DRG neurons. In spinal cord, p38 is activated between 5 h and 3 days after SNL and returns to baseline level within 5 days. Pretreatment with etanercept blocks p38 activation only in DRG, but not in spinal cord. These data indicate that phosphorylated p38 levels in spinal cord and DRG are transiently elevated after SNL treatment. In DRG, p38 activation is blocked by systemic TNF-α inhibition. Another report suggests the mechanism of TNF-α-induced pain in the line of the interaction with brain-derived neurotrophic factor (BDNF), which is thought to be a modulator of pain. Onda et al. [94] investigated the effect of infliximab, a chimeric monoclonal antibody to TNF-α, on induction of BDNF using an experimental herniated nucleus pulposus (NP) model. Application of NP induces a marked increase of BDNF immunoreactivity in number in the DRG neurons and within the superficial layer in the dorsal horn compared with the sham group. Intraperitoneal injection with infliximab reduces the BDNF induction in both DRG and spinal cord.
Possible involvement of P2X7 in chronic pain: IL-1β
Recently, accumulating evidence indicates the relationship between inflammatory cytokines including IL-1β and neuropathic pain [54, 64–68]. The expression of IL-1β is upregulated in the spinal cord of several rat mononeuropathy models [64–66]. Sweitzer et al. [64] investigated whether blocking the action of central IL-1β and TNF-α attenuates mechanical allodynia in a gender-specific manner in a rodent L5 spinal nerve transection model of neuropathic pain with/without glial activation. IL-1 receptor antagonist, not alone but in combination with soluble TNF-α receptor, decreases allodynia in a dose-dependent manner with glial activation remaining. At days 3 and 7 post-transection, the level of IL-6, but not IL-1β, in the L5 spinal cord of animals receiving daily IL-1 receptor antagonist in combination with soluble TNF-α receptor is significantly less than that of control animals. These findings further support a role for central IL-1β in the development and maintenance of neuropathic pain through induction of a proinflammatory cytokine cascade.
Di Virgilio, Ferrari and co-workers first reported that extracellular ATP triggers IL-1β release from LPS-treated microglia [95–97]. They confirmed that ATP is a powerful stimulus for IL-1β release from LPS-treated microglia and that IL-1β release occurs much earlier than the leakage of cytoplasmic markers. Sanz and Di Virgilio [97] examined the kinetics and mechanism of ATP-dependent IL-1β release from microglia. The addition of extracellular ATP to LPS-primed microglia causes a burst release of a large amount of processed IL-1β. ATP has no effect on the accumulation of intracellular pro-IL-1β in the absence of LPS. The optimal ATP concentration for IL-1β secretion is between 3 and 5 mM, but significant release can be observed at concentrations as low as 1 mM. At all ATP concentrations, the IL-1β release can be inhibited by increasing the extracellular K+ concentration. The ATP-dependent IL-1β release is also inhibited by the caspase inhibitors. Authors concluded that ATP triggers accelerated maturation and the release of intracellularly accumulated IL-1β by activating the IL-1β-converting enzyme/caspase 1 in mouse microglia. Extracellular ATP is the only endogenous compound known to cause a significant reduction in intracellular K+ and consequent release of IL-1β [97, 98]. Substantial evidence suggests a key role of P2X7 in the ATP-induced IL-1β release from LPS-primed microglia, i.e., (1) P2X7 antagonist oATP inhibits the release from microglia [96], (2) microglia lacking P2X7 does not release IL-1β after ATP stimulation [95]. Thus, it is suggested that an activation of P2X7 by ATP induces permitting movement of K+, Na+ and Ca2+ through the cell membrane and provokes the release of IL-1β from microglia.
Several cytokines have been reported to alter synaptic transmission in the CNS, including the spinal cord [99, 100]. The exogenous application of IL-1β enhances NMDA receptor-mediated Ca2+ responses via activating tyrosine protein kinase Src [101] which is known to enhance NMDA receptor activity in dorsal horn neurons [31, 102]. IL-1β also decreases GABAA receptor-mediated currents [103]. Mechanical and thermal hyperalgesia was absent in both inflammatory and neuropathic pain models in mice with a disrupted P2X7 gene, while normal nociceptive processing was preserved [35, 36], suggesting that the stimulation of P2X7 receptor expressed by satellite and Schwann cells causes the release of IL-1β and upregulation of nerve growth factor, resulting in the pain.
Thus, IL-1β released from activated microglia by activating P2X7 may also have modulatory effects on evoking neuropathic pain.
Involvement of P2X3 or P2X2/3 on DRG neurons in inflammatory pain
P2X-mediated nocifensive behavior is greatly enhanced in rats with inflammation at the hind paw [23]. A similar result was also obtained in humans [2, 104]. The stimulation of P2X receptors in an in-vitro skin-nerve preparation produced the excitation of C-mechanoheat polymodal nociceptors, which are enhanced in inflamed skin [105]. This evidence suggests that the levels of ATP in inflamed tissues are elevated [106, 107] and P2X receptors on peripheral nerve endings in inflamed areas would modulate pain processing. Indeed, P2X antagonists reduce hyperalgesia or tactile allodynia caused by complete Freund’s adjuvant (CFA) [22, 29, 108–110], an animal model of inflammatory pain. P2X3 antisense also reversed and prevented hyperalgesia in a CFA model [27, 111]. P2X3 and P2X2/3 antagonist A-317491 also showed a similar effect in a CFA model [22, 110, 112]. Mutant mice lacking P2X3 receptors, however, showed even enhanced, thermal hyperalgesia in a CFA model [21]. The discrepancy remains unresolved, but it might be due to some compensatory system resulting from P2X3 gene disruption. It is of particular interest to note that neither A-317491 nor P2X3 antisense reduces the heightened pain sensitivity by the carrageenan pain model [22, 111]. P2X3-knockout mice also display the carrageenan-induced hyperalgesia as do wild-type mice [21]. Since the inflammatory pain in the CFA model persists much longer than that in the carrageenan model, P2X receptors may play more important roles in chronic than acute inflammatory pain. This view is supported by evidence that persistent inflammation by CFA is accompanied by an upregulation of both P2X2 and P2X3 receptors in sensory neurons [113]. The increase in ATP responses in inflamed DRG neurons generates large depolarization [113]. Heightened responses via P2X receptors in DRG neurons in vitro are also observed in P2X receptors at the peripheral terminals in vivo. It was reported that activating P2X receptors on peripheral endings produces an activation of ERK in DRG neurons only under the situation in which peripheral tissues are inflamed [29]. The majority of activated ERK-positive DRG neurons express P2X3 receptors. The level of activated ERK is markedly enhanced by mechanical stimulation given to the inflamed hind paw, and, interestingly, the enhancement requires the activation of P2X receptors, presumably by endogenous ATP at the periphery. ATP is actually released from keratinocytes by mechanical stimulation [114]. Moreover, TNP-ATP, but not Ip5I, reduces CFA-induced mechanical allodynia [29], suggesting the predominant role of P2X2/3 receptors. This evidence suggeststhat persistent peripheral inflammation causes both quantitative and qualitative upregulation of P2X receptors in sensory neurons, which in turn leads to enhance responses of P2X receptors by released endogenous ATP, and thereby contributes to inflammatory pain hypersensitivity. The cellular mechanisms by which the expression and function of P2X receptors are upregulated in sensory neurons are still unknown, but several possibilities are considered. Since peripheral inflammation increases the levels of various inflammatory mediators in the inflamed area [32, 115], there may be an interaction between P2X receptors and these mediators that produces the functional upregulation of P2X receptors. Indeed, P2X-mediated responses are enhanced by substance P [116, 117], neurokinin B [118], prostaglandin E2 [23], protons [119] and bradykinin [117]. Some of the regulation is mediated through the phosphorylation of P2X receptors by protein kinases [117, 118]. For the upregulation of P2X receptor expression, growth factors could be candidates. Ramer et al. have shown that GDNF treatment increases P2X3 expression. Interestingly, NGF also increases the expression of P2X3 in DRG neurons and induces new expression in some DRG neurons. The fact that the NGF levels are dramatically elevated following peripheral inflammation suggests that de novo expression of P2X3 by NGF in trkA-positive DRG neurons could contribute to the enhanced ATP signaling via P2X receptors.
Involvement of P2X3 or P2X2/3 on DRG neurons in neuropathic pain
Peripheral nerve injury leads to intractable neuropathic pain that is often resistant to most current treatments [30–32]. Accumulating evidence has suggested that molecular and cellular alterations including P2X receptors in primary sensory neurons as well as in the dorsal horn after nerve injury have an important role in the pathogenesis of neuropathic pain. Transection of the sciatic nerve produces a marked reduction of P2X3-ir in injured DRG [121]. In contrast, an increase in the number of P2X3-ir-positive DRG neurons is seen following partial nerve injury by chronic constriction of the sciatic nerve [122] or of the inferior alveolar nerve [123], both of which are also models of neuropathic pain. The discrepancy in the change of P2X3 expression might be due to the type of nerve injury model, since a recent study using activating transcription factor 3, a neuronal injury maker, has shown that there is an increase in the expression of P2X3 in DRG neurons whose axons had been spared following partial nerve injury [33]. Besides P2X3, the regulation of other P2X receptors also changes following nerve injury. Messenger RNAs of P2X5 and P2X6 are increased and decreased, respectively, in the injured DRG [124]. Though P2X2 mRNA is unchanged, there is a marked increase in the number of P2X2-ir-positive neurons in the injured DRG [124]. These findings suggest posttranscriptional alterations as well as transcriptional changes, and raise the possibility that the composition of P2X subunits in P2X receptor may be changed in affected sensory neurons following nerve injury.
Molecularly reducing P2X3 expression in DRG neurons by P2X3 antisense or siRNA prevents the development of mechanical hypersensitivity by partial nerve injury of the sciatic nerve [27, 111, 125]. Interestingly, P2X3 antisense also reverses established neuropathic pain hypersensitivity [27, 111], which re-emerges within several days after the cessation of treatment with P2X3 antisense [111], suggesting that the P2X3 receptor also has an ongoing role. This is substantially supported by a recent finding that A-317491 reverses established allodynia and hyperalgesia after nerve injury [22, 110]. It is of note that reversing or preventing neuropathic pain behavior by P2X3 antagonists or antisense is observed not only in partial nerve injury models that are accompanied by the upregulation of P2X3 expression in DRG neurons [122] but also in spinal nerve injury models that decrease the expression of P2X3 receptors in L5 DRG neurons without affecting their expression in L4 DRG neurons [124, 126]. These findings indicate that P2X3 could contribute to the pathogenesis of neuropathic pain even if P2X3 expression in sensory neurons is not upregulated.
There are several possible mechanisms by which P2X receptors contribute to neuropathic pain. Based on evidence showing the presence of P2X3 receptors on peripheral nerve endings, and because ATP is released from peripheral tissues, including skin [5, 127, 128], P2X3 receptors in the periphery could be involved. This might, however, not be the case, since blocking P2X3 receptors on peripheral nerve endings by locally injection with A-317491 does not change the allodynia after partial nerve injury or spinal nerve injury [110]. However, a putative role of P2X3 receptor accumulation at the proximity of the injury site of sensory fibers [123, 129] can not be ruled out. In the dorsal horn, P2X receptors at central terminals could be involved since an intrathecal injection of A-317491 reverses allodynia [110]. Endogenous ATP may be released from central terminals of sensory fibers by excess stimulation after nerve injury [130], by dorsal horn interneurons [131], or by activated dorsal horn astrocytes [132]. The released ATP then activates P2X receptors at central terminals of the primary afferents, and thereby enhances the release of glutamate which results in an increase in the excitatory transmission in dorsal horn neurons.
Conclusion
Peripheral nerve injury leads to pathological changes in the spinal cord that cause the activation of spinal microglia. The activated microglia express P2X4, which can be stimulated by endogenous ATP, resulting in BDNF release and expression of neuropathic pain. Almost all currently known drugs for neuropathic pain were developed to target neurons, and these drugs do not exhibit adequate therapeutic effects in patients with neuropathic pain [133]. We expect that efforts to elucidate how P2X4R signaling in microglia causes neuropathic pain will provide us both with exciting insights into pain mechanisms and with clues to developing new therapeutic agents which may fundamentally change the management of intractable pain. P2X3 expressed by DRG neurons also plays an important role in inflammatory and neuropathic pain. P2X7 in microglia might be an essential molecule in producing cytokines, TNF-α and IL-1β which are involving in the chronic pain. These ATP receptors are thought to be new targets for drugs managing intractable chronic pain.
Abbreviations
- ADP:
-
adenosine 5–diphosphate
- ATP:
-
adenosine 5–triphosphate
- ATPγS:
-
adenosine 5–O-(3-thiotriphosphate)
- BDNF:
-
brain-derived neurotrophic factor
- BzATP:
-
2– and 3–O-(4-benzoylbenzoyl) adenosine 5–triphosphate
- [Ca2+]I:
-
Intracellular Ca2+ concentration
- CD11b:
-
cluster determinant 11b
- CNS:
-
central nervous system
- CR3:
-
complement receptor 3
- DRG:
-
dorsal root ganglion
- ERK:
-
extracellular signal-regulated protein kinase
- GDNF:
-
glial cell line-derived neurotrophic factor
- ICE:
-
IL-1β-converting enzyme
- IL-1β:
-
interleukin-1β
- IL-6:
-
interleukin-6
- INOS:
-
inducible nitric oxide synthase
- InsP3:
-
inositol 1, 4, 5-trisphosphate
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
lipopolysaccharide
- MAPK:
-
mitogen-activated protein kinase
- MEK:
-
mitogenactivated protein kinase kinase
- MHC:
-
histocompatibility complex
- NP:
-
nucleus pulposus
- oATP:
-
oxdized ATP
- PK11195:
-
[1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolineisoquinoline carboxamide]
- PTK:
-
protein tyrosine kinase
- PTX:
-
pertussis toxin
- SB203580:
-
4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)IH- imidazole
- SP600125:
-
anthra[1,9]pyrazol-6(2H)-one
- SNL:
-
spinal nerve ligation
- PPADS:
-
pyridoxalphosphate-6- azophenyl-2––disulphonic acid
- PKC:
-
protein kinase C
- PLC:
-
phospholipase C
- TNP-ATP:
-
2–3–O-(2,4,6-trinitrophenyl)adenosine 5–triphosphate
- TNF-α:
-
tumor necrosis factor-α
- U0126:
-
1,4-diamino-2,3-dicyano-1,4-bis[2-amino-phenylthio]butadiene
- UTP:
-
uridine 5–triphosphate
References
Bleehen T, Hobbiger F, Keele CA (1976) Identification of algogenic substances in human erythrocytes. J Physiol 262:131–49
Bleehen T, Keele CA (1977) Observations on the algogenic actions of adenosine compounds on the human blister base preparation. Pain 3:367–77
Chen CC, Akopian AN, Sivilotti L et al (1995) A P2X purinoceptor expressed by a subset of sensory neurons. Nature 377:428–31
Lewis C, Neidhart S, Holy C et al (1995) Coexpression of P2X2 and P2X3 receptor subunits can account for ATP- gated currents in sensory neurons. Nature 377:432–35
Burnstock G, Wood JN (1996) Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr Opin Neurobiol 6:526–32
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–92
Burnstock G (2000) P2X receptors in sensory neurones. Br J Anaesth 84:476–88
Cook SP, Rodland KD, McCleskey EW (1998) A memory for extracellular Ca2+ by speeding recovery of P2X receptors from desensitization. J Neurosci 18:9238–244
Ueno S, Tsuda M, Iwanaga T et al (1999) Cell type-specific ATP-activated responses in rat dorsal root ganglion neurons. Br J Pharmacol 126:429–36
Li C, Peoples RW, Lanthorn TH et al (1999) Distinct ATP-activated currents in different types of neurons dissociated from rat dorsal root ganglion. Neurosci Lett 263:57–0
Tsuda M, Koizumi S, Kita A et al (2000) Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J Neurosci 20:RC90
Chizh BA, Illes P (2001) P2X receptors and nociception. Pharmacol Rev 53:553–68
Dunn PM, Zhong Y, Burnstock G (2001) P2X receptors in peripheral neurons. Prog Neurobiol 65:107–34
Svichar N, Shmigol A, Verkhratsky A et al (1997) ATP induces Ca2+ release from IP3-sensitive Ca2+ stores exclusively in large DRG neurones. Neuroreport 8:1555–559
Koizumi S, Tsuda M, Shigemoto Y et al (2001) Characterization of P2Y receptors in cultured rat dorsal root ganglion neurons. Jpn J Pharmacol 85:149P
Sanada M, Yasuda H, Omatsu-Kanbe M et al (2002) Increase in intracellular Ca(2+) and calcitonin gene-related peptide release through metabotropic P2Y receptors in rat dorsal root ganglion neurons. Neuroscience 111:413–22
Tominaga M, Wada M, Masu M (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98:6951–956
Molliver DC, Cook SP, Carlsten JA et al (2002) ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2. Eur J Neurosci 16:1850–860
Moriyama T, Iida T, Kobayashi K et al (2003) Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci 23:6058–062
Cockayne DA, Hamilton SG, Zhu QM et al (2000) Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature 407:1011–015
Souslova V, Cesare P, Ding Y et al (2000) Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature 407:1015–017
Jarvis MF, Burgard EC, McGaraughty S et al (2002) A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA 99:17179–7184
Hamilton SG, Wade A, McMahon SB (1999) The effects of inflammation and inflammatory mediators on nociceptive behaviour induced by ATP analogues in the rat. Br J Pharmacol 126:326–32
Bland-Ward PA, Humphrey PP (1997) Acute nociception mediated by hindpaw P2X receptor activation in the rat. Br J Pharmacol 122:365–71
Tsuda M, Ueno S, Inoue K (1999) In vivo pathway of thermal hyperalgesia by intrathecal administration of alpha,beta-methylene ATP in mouse spinal cord: involvement of the glutamate-NMDA receptor system. Br J Pharmacol 127:449–56
Dorn G, Abdel’Al S, Natt FJ et al (2001) Specific inhibition of the rat ligand-gated ion channel P2X3 function via methoxyethoxy-modified phosphorothioated antisense oligonucleotides. Antisense Nucleic Acid Drug Dev 11:165–74
Barclay J, Patel S, Dorn G et al (2002) Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci 22:8139–147
Tsuda M, Shigemoto-Mogami Y, Ueno S et al (2002) Downregulation of P2X3 receptor-dependent sensory functions in A/J inbred mouse strain. Eur J Neurosci 15:1444–450
Dai Y, Fukuoka T, Wang H et al (2004) Contribution of sensitized P2X receptors in inflamed tissue to the mechanical hypersensitivity revealed by phosphorylated ERK in DRG neurons. Pain 108:258–66
Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353:1959–964
Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science 288:1765–769
Scholz J, Woolf CJ (2002) Can we conquer pain? Nat Neurosci 5:1062–067
Tsuzuki K, Kondo E, Fukuoka T et al (2001) Differential regulation of P2X(3) mRNA expression by peripheral nerve injury in intact and injured neurons in the rat sensory ganglia. Pain 91:351–60
Kennedy C, Assis TS, Currie AJ et al (2003) Crossing the pain barrier: P2 receptors as targets for novel analgesics. J Physiol 553:683–94
Casula MA, Chessell IP, Bountra C, Yiangou Y, Birch R, Anand P (2004) Increase of purinergic receptor P2X7 in injured human nerves anddorsal root ganglia. J Neurol Neurosurg Psychiatry 75:1227–231
Chessell P, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P et al (2005) Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114:386–96
Tsuda M, Shigemoto-Mogami Y, Koizumi S et al (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424:778–83
Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017–021
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–18
Stoll G, Jander S (1999) The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 58:233–47
Perry VH (1994) Modulation of microglia phenotype. Neuropathol Appl Neurobiol 20:177–84
Wall PD, Devor M, Inbal R et al (1979) Autotomy following peripheral nerve lesions: experimental anaesthesia dolorosa. Pain 7:103–11
Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33:87–07
Seltzer Z, Dubner R, Shir Y (1990) A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43:205–18
Kim SH, Chung JM (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–63
Decosterd I, Woolf CJ (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87:149–58
Eriksson NP, Persson JK, Svensson M et al (1993) A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp Brain Res 96:19–7
Colburn RW, DeLeo JA, Rickman AJ et al (1997) Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol 79:163–75
Coyle DE (1998) Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia 23:75–3
Colburn RW, Rickman AJ, DeLeo JA (1999) The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol 157:289–04
Stuesse SL, Cruce WL, Lovell JA et al (2000) Microglial proliferation in the spinal cord of aged rats with a sciatic nerve injury. Neurosci Lett 287:121–24
Gehrmann J, Banati RB (1995) Microglial turnover in the injured CNS: activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J Neuropathol Exp Neurol 54:680–88
Liu L, Tornqvist E, Mattsson P et al (1995) Complement and clusterin in the spinal cord dorsal horn and gracile nucleus following sciatic nerve injury in the adult rat. Neuroscience 68:167–79
Watkins LR, Milligan ED, Maier SF (2001) Glial activation: a driving force for pathological pain. Trends Neurosci 24:450–55
Surprenant A, Rassendren F, Kawashima E et al (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272:735–38
Khakh BS, Burnstock G, Kennedy C et al (2001) International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev 53:107–18
Virginio C, Robertson G, Surprenant A et al (1998) Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol Pharmacol 53:969–73
Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y (2003) Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424:938–42
Mannion RJ et al (1999) Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA 96:9385–390
Heppenstall PA, Lewin GR (2001) BDNF but not NT-4 is required for normal flexion reflex plasticity and function. Proc Natl Acad Sci USA 98:8107–112
Thompson SW, Bennett DL, Kerr BJ et al (1999) Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci USA 96:7714–718
Rivera C (2002) BDNF-induced TrkB activation down-regulates the K+-Cl– cotransporter KCC2 and impairs neuronal Cl– extrusion. J Cell Biol 159:747–52
Gravel C, Gotz R, Lorrain A, Sendtner M (1997) Adenoviral gene transfer of ciliary neurotrophic factor and brain-derived neurotrophic factor leads to longterm survival of axotomized motor neurons. Nature Med 3:765–70
Sweitzer S, Martin D, DeLeo JA (2001) Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience 103:529–39
Winkelstein BA, Rutkowski MD, Sweitzer SM et al (2001) Nerve injury proximal or distal to the DRG induces similar spinal glial activation and selective cytokine expression but differential behavioral responses to pharmacologic treatment. J Comp Neurol 439:127–39
Raghavendra V, Tanga F, DeLeo JA (2003) Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 306:624–30
Robertson B, Xu XJ, Hao JX et al (1997) Interferon-gamma receptors in nociceptive pathways: role in neuropathic pain-related behaviour. Neuroreport 8:1311–316
DeLeo JA, Yezierski RP (2001) The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 90:1–
Wagner R, Myers RR (1996) Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. NeuroReport 7:2897–901
Sommer C, Schäfers M (1998) Painful mononeuropathy in C57BL/Wld mice with delayed Wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res 784:154–62
Sommer C, Marziniak M, Myers RR (1998) The effect of thalidomide treatment on vascular pathology and hyperalgesia caused by chronic constriction injury of rat nerve. Pain 74:83–1
Sorkin LS, Doom CM (2000) Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst 5:96–00
Schäfers M, Geis C, Brors D, Yaksh TL, Sommer C (2002) Anterograde transport of tumor necrosis factor-alpha in the intact and injured rat sciatic nerve. J Neurosci 22:536–45
Schäfers M, Lee DH, Brors D, Yaksh TL, Sorkin LS (2003) Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci 23:3028–038
Hashizume H, DeLeo, JA, Colburn RW, Weinstein JN (2000) Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine 25:1206–217
Sommer C, Schäfers M, Marziniak M, Toyka KV (2001) Etanercept reduces hyperalgesia in experimental painful neuropathy. J Peripher Nerv Syst 6:67–2
Sommer C, Lindenlaub T, Teuteberg P, Schäfers M, Hartung T, Toyka KV (2001) Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res 913:86–9
Homma Y, Brull SJ, Zhang JM (2002) A comparison of chronic pain behavior following local application of tumor necrosis factor-alpha to the normal and mechanically compressed lumbar ganglia in the rat. Pain 95:239–46
Sawada M, Hara N, Maeno T (1990) Extracellular tumor necrosis factor induces a decreased K+ conductance in an identified neuron of Aplysia kurodai. Neurosci Lett 115:219–25
Soliven B, Albert J (1992) Tumor necrosis factor modulates Ca2+ currents in cultured sympathetic neurons. J Neurosci 12:2665–671
Sorkin LS, Xiao WH, Wagner R, Myers RR (1997) Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience 81:255–62
Furukawa K, Mattson MP (1998) The transcription factor NF-kappa B mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor-alpha in hippocampal neurons. J Neurochem 70:1876–886
Junger H, Sorkin LS (2000) Nociceptive and inflammatory effects of subcutaneous TNF-α. Pain 85:145–51
Diem R, Meyer R, Weishaupt JH, Bahr M (2001) Reduction of potassium currents and phosphatidylinositol 3-kinase-dependent Akt phosphorylation by tumor necrosis factor-α rescues axotomized retinal ganglion cells from retrograde cell death in vivo. J Neurosci 21:2058–066
Leem JG, Bove GM (2002) Mid-axonal tumor necrosis factor-alpha induces ectopic activity in a subset of slowly conducting cutaneous and deep afferent neurons. J Pain 3:45–9
Hide I, Tanaka M, Inoue A et al (2000) Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem 75:965–72
Suzuki T, Hide I, Ido K et al (2004) Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci 24:1–
Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ,et al (2002) TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology 42:93–06
Ji R, Samad T, Jin S, Schmoll R, Woolf C (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 3:57–8
Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S (2003) Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 23:1026–040
Jin SX, Zhuang ZY, Woolf CJ et al (2003) p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 23:4017–022
Tsuda M, Mizokoshi A, Shigemoto-Mogami Y et al (2004) Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 45:89–5
Schäfers M, Svensson CI, Sommer C, Sorkin LS (2003) Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 23:2517–521
Onda A, Murata Y, Rydevik B, Larsson K, Kikuchi S, Olmarker K (2004) Infliximab attenuates immunoreactivity of brain-derived neurotrophic factor in a rat model of herniated nucleus pulposus. Spine 29:1857–861
Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F (1996) Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol 156:1531–539
Ferrari D, Chiozzi P, Falzoni S, Hanau S, Di Virgilio F (1997) Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med 185:579–82
Sanz JM, Di Virgilio F (2000) Kinetics and mechanism of ATP-dependent IL-1 beta release from microglial cells. J Immunol 164:4893–898
Perregaux D, Gabel CA (1994) Interleukin-1 maturation and release in response to ATP and nigericin. J Biol Chem 269:15195–5203
Kerr BJ, Bradbury EJ, Bennett DL et al (1999) Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci 19:5138–148
Vikman KS, Hill RH, Backstrom E et al (2003) Interferon-gamma induces characteristics of central sensitization in spinal dorsal horn neurons in vitro. Pain 106:241–51
Viviani B, Bartesaghi S, Gardoni F et al (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23:8692–700
Yu XM, Askalan R, Keil GJ 2nd et al (1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275:674–78
Wang S, Cheng Q, Malik S et al (2000) Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther 292:497–04
Hamilton SG, Warburton J, Bhattacharjee A, Ward J, McMahon SB (2000) ATP in human skin elicits a dose-related pain response which is potentiated under conditions of hyperalgesia. Brain 123:1238–246
Hamilton SG, McMahon SB, Lewin GR (2001) Selective activation of nociceptors by P2X receptor agonists in normal and inflamed rat skin. J Physiol 534:437–45
Ryan LM, Rachow JW, McCarty DJ (1991) Synovial fluid ATP: a potential substrate for the production of inorganic pyrophosphate. J Rheumatol 18:716–20
Park W, Masuda I, Cardenal-Escarcena A, Palmer DL, McCarty DJ (1996) Inorganic pyrophosphate generation from adenosine triphosphate by cell-free human synovial fluid. J Rheumatol 23:665–71
Dell’Antonio G, Quattrini A, Cin ED, Fulgenzi A, Ferrero ME (2002) Relief of inflammatory pain in rats by local use of the selective P2X7 ATP receptor inhibitor, oxidized ATP. Arthritis Rheum 46:3378–385
Dell’Antonio G, Quattrini A, Dal Cin E, Fulgenzi A, Ferrero ME (2002) Antinociceptive effect of a new P2Z/P2X7 antagonist, oxidized ATP, in arthritic rats. Neurosci Lett 327:87–0
McGaraughty S, Wismer CT, Zhu CZ, Mikusa J, Honore P, Chu KL, Lee CH, Faltynek CR, Jarvis MF (2003) Effects of A-317491, a novel and selective P2X3/P2X2/3 receptor antagonist, on neuropathic, inflammatory and chemogenic nociception following intrathecal and intraplantar administration. Br J Pharmacol 140:1381–388
Honore P, Kage K, Mikusa J et al (2002) Analgesic profile of intrathecal P2X(3) antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain 99:11–9
Wu G, Whiteside GT, Lee G, Nolan S, Niosi M, Pearson MS, Ilyin VI (2004) A-317491, a selective P2X3/P2X2/3 receptor antagonist, reverses inflammatory mechanical hyperalgesia through action at peripheral receptors in rats. Eur J Pharmacol 504:45–3
Xu GY, Huang LY (2002) Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci 22:93–02
Koizumi S, Fujishita K, Inoue K, Shigemoto-Mogami Y, Tsuda M (2004) Ca2+ waves in keratinocytes are transmitted to sensory neurons: the involvement of extracellular ATP and P2Y2 receptor activation. Biochem J 380:329–38
Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413:203–10
Hu HZ, Li ZW (1996) Substance P potentiates ATP-activated currents in rat primary sensory neurons. Brain Res 739:163–68
Paukert M, Osteroth R, Geisler HS, Brandle U, Glowatzki E, Ruppersberg JP, Grunder S (2001) Inflammatory mediators potentiate ATP-gated channels through the P2X3 subunit. J Biol Chem 276:21077–1082
Wang MJ, Xiong SH, Li ZW (2001) Neurokinin B potentiates ATP-activated currents in rat DRG neurons. Brain Res 923:157–62
Li C, Peoples RW, Weight FF (1997) Enhancement of ATP-activated current by protons in dorsal root ganglion neurons. Pflugers Arch 433:446–54
Ramer MS, Bradbury EJ, McMahon SB (2001) Nerve growth factor induces P2X3 expression in sensory neurons. J Neurochem 77:864–75
Bradbury EJ, Burnstock G, McMahon SB (1998) The expression of P2X3 purinoreceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Mol Cell Neurosci 12:256–68
Novakovic SD, Kassotakis LC, Oglesby IB, Smith JA, Eglen RM, Ford AP, Hunter JC (1999) Immunocytochemical localization of P2X3 purinoceptors in sensory neurons in naive rats and following neuropathic injury. Pain 80:273–82
Eriksson J, Bongenhielm U, Kidd E, Matthews B, Fried K (1998) Distribution of P2X3 receptors in the rat trigeminal ganglion after inferior alveolar nerve injury. Neurosci Lett 254:37–0
Kim C, Chung JM, Chung K (2003) Changes in the gene expression of six subtypes of P2X receptors in rat dorsal root ganglion after spinal nerve ligation. Neurosci Lett 337:81–4
Dorn G, Patel S, Wotherspoon G, Hemmings-Mieszczak M, Barclay J, Natt FJ, Martin P, Bevan S, Fox A, Ganju P, Wishart W, Hall J (2004) siRNA relieves chronic neuropathic pain. Nucleic Acids Res 32:e49
Kage K, Niforatos W, Zhu CZ, Lynch KJ, Honore P, Jarvis MF (2002) Alteration of dorsal root ganglion P2X3 receptor expression and function following spinal nerve ligation in the rat. Exp Brain Res 147:511–19
Cook SP, McCleskey EW (2002) Cell damage excites nociceptors through release of cytosolic ATP. Pain 95:41–7
Koizumi S, Fujishita K, Inoue K et al (2004) Ca2+ waves in keratinocytes are transmitted to sensory neurons: the involvement of extracellular ATP and P2Y2 receptor activation. Biochem J 380:329–38
Vulchanova L, Riedl MS, Shuster SJ, Stone LS, Hargreaves KM, Buell G, Surprenant A, North RA, Elde R (1998) P2X3 is expressed by DRG neurons that terminate in inner lamina II. Eur J Neurosci 10:3470–478
Nakatsuka T, Gu JG (2001) ATP P2X receptor-mediated enhancement of glutamate release and evoked EPSCs in dorsal horn neurons of the rat spinal cord. J Neurosci 21:6522–531
Jo YH, Schlichter R (1999) Synaptic corelease of ATP and GABA in cultured spinal neurons. Nat Neurosci 2:241–45
Fam SR, Gallagher CJ, Salter MW (2000) P2Y1 purinoceptor-mediated Ca2+ signaling and Ca2+ wave propagation in dorsal spinal cord astrocytes. J Neurosci 20:2800–808
Watkins LR, Maier SF (2003) GLIA: A novel drug discovery target for clinical pain. Nat Rev Drug Discov 2:973–85
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Inoue, K. P2 receptors and chronic pain. Purinergic Signalling 3, 135–144 (2007). https://doi.org/10.1007/s11302-006-9045-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-006-9045-8