
Abstract
Within the last 15 years, at least 8 different G protein-coupled P2Y receptors have been characterized. These mediate slow metabotropic effects of nucleotides in neurons as well as non-neural cells, as opposed to the fast ionotropic effects which are mediated by P2X receptors. One class of effector systems regulated by various G protein-coupled receptors are voltage-gated and ligand-gated ion channels. This review summarizes the current knowledge about the modulation of such neuronal ion channels via P2Y receptors. The regulated proteins include voltage-gated Ca2+ and K+ channels, as well as N-methyl-d-aspartate, vanilloid, and P2X receptors, and the regulating entities include most of the known P2Y receptor subtypes. The functional consequences of the modulation of ion channels by nucleotides acting at pre- or postsynaptic P2Y receptors are changes in the strength of synaptic transmission. Accordingly, ATP and related nucleotides may act not only as fast transmitters (via P2X receptors) in the nervous system, but also as neuromodulators (via P2Y receptors). Hence, nucleotides are as universal transmitters as, for instance, acetylcholine, glutamate, or γ-aminobutyric acid.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many functional characteristics of neurons are similar or even identical to those of other cells, but there is one fundamental difference: The primary functions of neuronal cells are to receive, modify, and transmit messages. This flow of information occurs within one neuron as well as between neurons. Whether the neuronal communication is intra- or intercellular, it is always dependent on electrical activity which is provided by ligand- as well as voltage-gated ion channels. With respect to electrical properties, different neurons are by far not equal but rather characterized by prototypical synaptic responses that are governed by the expression of a certain set of ion channels. Even within one nerve cell, the electrical properties are not fixed, but may change with time depending on the presence of neurotransmitters, hormones, or growth factors. The basis for changes in the electrical responsiveness of a neuron are alterations in the opening and closure of ion channels. Such changes are in many cases triggered by neurotransmitters that act via heptahelical transmembrane receptors typically linked to heterotrimeric G proteins [1].
Adenine and uridine nucleotides are present in all types of cells and are released in response to various stimuli [2]. Once in the extracellular space, they activate one or more of at least 15 different membrane proteins which are categorized as P2 receptors. Alternatively, they are converted to other nucleotides or degraded down to nucleosides by different ectoenzymes [3]. The receptors for nucleosides are categorized as P1 receptors. Receptors for nucleotides comprise ionotropic P2X and metabotropic P2Y receptors. P2X receptors are ATP-gated cation channels composed of presumably three subunits out of a repertoire of at least seven different proteins which all have two presumed transmembrane domains with large extracellular loops and intracellular N- and C-termini [4, 5]. P2Y receptors, in contrast, are characterized by seven putative transmembrane domains typical of G protein-coupled receptors. Currently, at least eight different subtypes have been identified (P2Y1, 2, 4, 6, 11, 12, 13, 14; [4, 6–8]. Thus, with respect to the modulation of ion channels, receptors for nucleotides may represent the regulated entity in the case of P2X receptors, and they may also be the regulating entity in the case of P2Y receptors. This review focuses on the interactions between P2Y receptors and both, ligand- as well as voltage-gated, ion channels and therefore deals with P2X receptors in only a very few instances.
Pharmacological and functional characteristics of P2Y receptors
A considerable number of heptahelical receptors that can be activated by extracellular nucleotides have been cloned from various species. According to the structural classification of P2 receptors they were categorized as P2Y receptors and numbered in chronologic order which led to the description of P2Y1 through P2Y15 receptors [9]. However, some of the receptors provided with P2Y receptor numbers were subsequently identified as species homologues of other P2Y receptors or as members of other families of G protein-coupled receptors [6]. The presumed P2Y15 receptor is also activated by α-ketoglutarate [10] and may thus not be a true member of the P2Y family. Accordingly, only eight different mammalian subtypes are currently viewed as members of the P2Y family, namely P2Y1, 2, 4, 6, 11, 12, 13, 14 [4, 6–8].
These P2Y receptors are activated not only by ATP, but also by other naturally occurring nucleotides or nucleotide sugars, such as ADP, UTP, UDP, and UDP glucose [4, 6–8]. At P2Y1 receptors of various species, the rank order of agonist potency is 2-MeSADPQ ≥ 2-MeSATP > ADP ≥ ATP with uridine nucleotides being inactive. At P2Y2 receptors, ATP and UTP are equipotent agonists, and ADP, UDP, or 2-methylthio derivatives have weak or no activity. P2Y4 receptors are activated by UTP, and the rat and mouse receptors are also activated by ATP, whereas the human receptors are antagonized by ATP [11]. At P2Y6 receptors, UDP is the most potent agonist and ADP, ATP or UTP are, if at all, only weak agonists. Human P2Y11 receptors show the following rank order of agonist potency: ATP ≥ 2-MeSATP > ADP, and the observed agonistic activity of UTP depends on the signalling cascade that is activated by the receptor [12]. The nucleotide selectivity of canine P2Y11 receptors is 2-MeSATP > ADP > ATP [13]. At P2Y12 receptors, 2-MesADP is much more potent as an agonist than ADP, and the efficacy of ATP is species-dependent with high intrinsic activity at rat but not at human receptors [14, 15]. P2Y13 receptors are activated by 2-MesADP, ADP, and ATP, but the rank order of agonist potency is different for human, murine, and rat receptors [16–18]. The P2Y14 receptor is activated by various UDP sugars, but not by adenine or uridine nucleotides [8, 19].
According to the agonist profiles summarized above, P2Y receptors can be characterized as receptors for purines (P2Y1, 11, 12, 13), for pyrimidines (P2Y6, 14), or for both types of nucleotides (P2Y2, 4). In parallel, these receptors can be classified as receptors preferring nucleoside tri-(P2Y2, 4, 11) or diphosphates (P2Y1, 6, 12, 13, 14). This latter categorization appears particularly important from a functional point of view, as extracellular nucleotides are released from a large variety of cells and rapidly converted by ectoenzymes [3]. The interconversion between different extracellular nucleotides is also a factor that needs to be taken into consideration, when P2Y receptors are characterized by the application of various nucleotides. For instance, the triphosphate sensitivity of P2Y2 and 4 receptors can only be demonstrated unequivocally, if the conversion of nucleoside diphosphates to triphosphates is prevented [20]. Given these experimental difficulties with endogenous and exogenous agonistic nucleotides, the characterization of P2Y receptors should preferably rely on the use of specific antagonists. A considerable number of compounds have been found to block P2Y receptors, but unfortunately the majority of antagonists is not sufficiently selective. For instance, the most widely used P2 receptor antagonists suramin and reactive blue 2 block not only a large number of P2X and P2Y receptors, but also unrelated receptors, such as NMDA receptors [21] and anion channels [22]. Nevertheless, for some P2Y receptor subtypes selective antagonists have been identified or developed. For instance, adenosine-2′-phosphate-5′-phosphate (A2P5P) and adenosine-3′-phosphate-5′-phosphate (A3P5P) were found to be partial agonists with low intrinsic activity at P2Y1 receptors [23]. Derivatives thereof, such as N 6-methyldeoxyadenosine 3′,5′-biphosphate (MRS2179) or 2-chloro-N 6-methyldeoxyadenosine 3′,5′-biphosphate (MRS 2216) are selective and competitive antagonist at P2Y1 receptors with nanomolar affinity [24]. The P2Y12 receptor has been identified as the target of metabolites of the well-known antithrombotic drugs ticlopidine and clopidogrel [25]. ATP derivatives, such as AR-C69931MX, are also antagonists with high affinity at this receptor subtype and were developed for clinical use in patients with acute coronary syndromes [26]. Unfortunately, this latter agent is not absolutely selective for P2Y12, but also blocks P2Y13 receptors. However, at P2Y12 this agent is a competitive, and at P2Y13 a non-competitive, antagonist [17]. Recently, insurmountable antagonists with nanomolar affinities for P2Y6 receptors, but no activity at P2Y1,2,4 and P2Y11 receptors, such as 1,4-di-[(3-isothiocyanato phenyl)-thioureido]butane (MRS 2578), have been developed [27].
P2Y1, 2, 6, 13, and P2Y14 are widely expressed in a variety of tissues including the nervous system, whereas P2Y4 and 11 show a restricted expression pattern that excludes neuronal tissues [4, 9, 19, 28]. P2Y1 mRNA and protein is found at high levels in many regions of the central nervous system including the cerebral and cerebellar cortices, hippocampus, caudate nucleus, putamen, globus pallidus and midbrain [29–31]. In in situ hybridizations of brain sections, the expression pattern of P2Y12 was consistent with a predominantly glial localization [25, 32]. P2Y13 mRNA was also detected in different brain regions, such as the thalamus, caudate nucleus, substantia nigra, hippocampus and cerebellum [28].
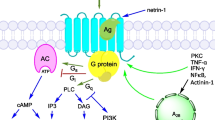
In heterologous expression systems, all P2Y receptor subtypes were found to mediate increases in inositol phosphates (IPs) or in intracellular Ca2+, thus indicating that they are coupled to phospholipase C (PLC) [4, 9, 28, 32]. The P2Y receptor-mediated increases in IPs did not involve pertussis toxin-sensitive G proteins in the cases of P2Y1, 6 and 11, but were to various degrees reduced by that bacterial toxin in the cases of P2Y2 and P2Y4 [4]. With P2Y12, 13 and P2Y14 receptors, nucleotide-dependent increases in IPs were only detected when the receptors were co-expressed together with either Gα16 or chimaeric G protein α subunits, and the actions mediated by these receptors were pertussis toxin-sensitive and thus mediated by inhibitory G proteins [19, 28]. P2Y11 mediates increases in cyclic AMP in addition to the rises in IPs [9, 33], and the coupling of this receptor to different effector systems displays different agonist sensitivities [12]. P2Y12 and P2Y13 mediate a pertussis toxin-sensitive inhibition of adenylyl cyclase [25, 28], and in pertussis toxin-treated cells, P2Y13 mediates a stimulation of adenylyl cyclase [28].
Regulation of neuronal ion channels via G protein-coupled receptors
Ion channels and G protein-coupled receptors of neurons are encoded by several hundred genes, and the list is still growing. Accordingly, it is impossible to summarize all the interactions between them, and this chapter highlights only a few examples which are important for the understanding of mechanisms that may link P2Y receptors to various ion channels. The ion channels that will be described below include ligand-gated ion channels as well as voltage-gated Na+, Ca2+ and K+ channels.
Currently, at least nine different types of pore forming subunits of voltage-gated Na+ channels are known and they contain several sites that may become phosphorylated by protein kinases A and C. On a functional level, phosphorylation by both kinases leads to reduced Na+ currents [34]. In accordance with this, D1-like receptors were found to reduce Na+ currents in hippocampal neurons via protein kinase A [35], and this effect was enhanced at hyperpolarized membrane potentials and by co-activation of protein kinase C [36]. Moreover, activation of muscarinic receptors reduced Na+ currents via protein kinase C in hippocampal [37] and neocortical neurons [38]. A similar effect was observed in neurons of the prefrontal cortex when 5-HT2 receptors were activated [39]. In sensory neurons, however, hyperalgesic agents increase currents through tetrodotoxin-resistant Na+ channels [40], and this effect was again mediated by protein kinase A [41]. Thus, several types of G protein-coupled receptors were found to modulate Na+ channels and increases as well as decreases in currents were observed depending on the types of neurons that were investigated.
Voltage-activated Ca2+ channels are classified by the genes encoding the pore forming a1 subunits, and one can discern between three subfamilies named CaV1 to CaV3. Members of all three families contribute to the voltage-gated Ca2+ currents found in neurons. Channels formed by CaV1 proteins mediate L-type currents, those containing, CaV2.1 to CaV2.3 mediate P/Q-, N- and R-type currents, and those harbouring CaV3 subunits mediate T-type Ca2+ currents [42]. While channels providing N- and P/Q-type currents are involved in excitation secretion coupling in most nerve terminals, the other channels are rather found at somatodendritic regions of neurons [42]. Modulation via G protein-coupled receptors has been described for an exhaustive number of neurotransmitters and for all types of voltage-gated Ca2+ currents [43–46]. The modulation of L-type currents is either facilitatory or inhibitory and is in most cases mediated by diffusible second messengers and protein kinases [45]. In contrast, the modulation of N and P/Q-type channels is almost exclusively inhibitory and is frequently independent of diffusible second messengers and protein kinases [43, 44], although there are some exceptions to this rule [47, 48]. The pathway that excludes diffusible second messengers is membrane-delimited and leads to a voltage-dependent inhibition of currents as revealed by the fact that large depolarisations attenuate the inhibition. This inhibition is in most, but not all, cases abolished by pertussis toxin [43], and is based on a direct interaction between G protein βγ subunits and Ca2+ channel proteins [44, 46]. Pathways involving the synthesis of diffusible second messengers most commonly lead to a voltage-independent reduction of Ca2+ currents and have been reported to involve a subunits of the Gq/11 protein family [49, 50].
The superfamily of voltage-dependent K+ channels comprises many more members than those of Na+ or Ca2+ channels, and the K+ channels are, in addition, very heterogeneous [51]. Although quite a number of these various K+ channels were reported to be modulated by neurotransmitters, the most intensively studied examples of K+ channel regulation via G protein-coupled receptors are inward rectifier (Kir) channels and KCNQ channels which are now classified as KV7 family [51]. In a variety of neurons, several neurotransmitters were found to cause hyperpolarizations by activating inwardly rectifying K+ currents via receptors coupled to pertussis toxin-sensitive G proteins. These effects were found to involve proteins of the Kir3 family and G protein βγ subunits [52]. However, the regulation of G protein-coupled inwardly rectifying K+ (GIRK) channels depends not only on βγ subunits, but also on other proteins and second messengers. G Protein α subunits may act as donors for βγ, on one hand, and directly block GIRK channels [53], on the other hand. Furthermore, the kinetics of the gating of GIRKs is determined by all three parameters, receptor type, G protein α, and G protein βγ subunits [54]. In addition, regulators of G protein signalling also determine the kinetics of GIRK activation [55]. Moreover, GIRK channels are activated by phosphatidylinositol 4,5-bisphosphate (PIP2) [56], and the levels of PIP2 are also regulated via G protein-coupled receptors and PLC [57]. As a consequence, activation of receptors linked to Gq proteins may also contribute to the regulation of GIRK channels [58]. Finally, activation of Gs coupled receptors was reported to enhance currents through GIRK channels [59].
A number of neurotransmitters were found to depolarize neurons by reducing M-type K+ currents (I M) which are believed to be mediated by KCNQ channels [60]. These ion channels are opened in the subthreshold voltage range for action potentials and become completely activated when neurons are further depolarized. Hence, activated KCNQ channels keep neurons polarized, and closure of these ion channels causes depolarization and increased action potential discharge [61, 62]. The inhibition of I M via M1 muscarinic acetylcholine receptors involves αq subunits of heterotrimeric GTP binding proteins [63], and a reduction in PIP2 through an activation of phospholipase C [64]. The inhibition of I M via bradykinin B2 receptors, in contrast, involves G proteins of the Gq family, most likely G11, and rather α then βγ subunits [50, 63, 65]. Via these G proteins, phospholipase C-β4 is stimulated [66] to mediate the synthesis of IP3, which then causes liberation of Ca2+ from intracellular stores [67]. Cytosolic Ca2+ concentrations in the sub- to low micromolar range directly block KM channels [68].
The nicotinic acetylcholine receptor as a prototypical ligand-gated ion channel has been shown to be modulated via a variety of G protein-coupled receptors, and receptor activation was found to either enhance or reduce the function of that ion channel [69–73]. The inhibition of currents through nicotinic acetylcholine receptor was suggested to be mediated by an inhibition of proteinkinase A-dependent phosphorylation [73]. The enhancement of these currents involved pertussis toxin-sensitive G proteins, but not altered phosphorylation [72]. A direct interaction between G protein subunits and ligand-gated ion channels has been demonstrated for glycine receptors. Currents through these ion channels were enhanced by free G protein βγ subunits [74]. Like nicotinic acetylcholine receptors, glycine receptors were found to be inhibited by prostaglandin E2, and this effect involves proteinkinase A-mediated phosphorylation [75]. Another ligand-gated ion channel, the glutamatergic NMDA receptor, is also regulated by phosphorylation secondary to the activation of G proteincoupled receptors. In that case, the activation of muscarinic acetylcholine receptors enhanced NMDA receptor currents via protein kinase C and non-receptor tyrosine kinase [76]. However, D2-like receptors reduce currents through NMDA receptors via transactivation of receptor tyrosine kinases [77]. In addition, D1 receptors may also directly interact with NMDA receptors [78]. For the inhibitory transmitter GABA, a direct interaction of the ionotropic GABAA receptors and the metabotropic GABAB receptor has been reported to regulate receptor trafficking [79]. Hence, a multitude of mechanisms provide functional links between G protein-coupled receptors and various ligand-gated ion channels.
Regulation of Na+ channels via P2Y receptors
ATP was found to enhance or inhibit tetrodotoxin-sensitive as well as -insensitive voltage-gated sodium channels in rat dorsal root ganglion neurons [80] and in a neuronal hippocampal cell line [81]. However, these effects did not appear to involve G proteins and were rather mediated by a direct interaction of ATP with the sodium channel. No additional evidence for a modulation of neuronal Na+ channels via P2Y receptor could be found in the literature.
Regulation of Ca2+ channels via P2Y receptors
Native receptors
Since Dunlap and Fischbach [82, 83] first described transmitter-induced inhibition of voltage-gated calcium channels, a plethora of other neurotransmitters and hormones that exert their actions via G protein-coupled receptors have been shown to inhibit native Ca2+ currents (see above). First direct evidence for a nucleotide regulation of voltage-gated calcium channels was presented by Diverse-Pierluissi et al. [84], who showed that ATP and ADP, but not UTP, induce a pertussis toxin-sensitive inhibition of voltage-gated Ca2+ currents in bovine adrenal chromaffin cells. Subsequent studies by several other groups [85–87] corroborated these results, and further showed that this reduction of calcium current amplitudes was voltage-dependent, antagonized by reactive blue 2 and due to an inhibition of N- and P/Q-type calcium channels. Similar results were obtained in rat adrenal chromaffin cells, where ATP also inhibited voltage-gated Ca2+ currents in a PTX-sensitive manner and this effect was also antagonized by reactive blue 2 [88]. Another ATP-induced inhibition of Ca2+ currents, which was PTX-insensitive and voltage-dependent, was observed in frog sympathetic neurons [89]. In NG108-15 mouse neuroblastoma × rat glioma cells, not only adenine nucleotides, but also UTP and UDP, inhibited whole-cell Ca2+ currents [90], with the uridine nucleotides being even more potent agonists than the adenine nucleotides. In this case, not only an inhibition of N-type channels was responsible for the reduction of voltage-gated Ca2+ currents, but also L-type calcium channels were inhibited. Interestingly, only the N-type channel inhibition was PTX-sensitive, while currents through L-type channels were still modulated after PTX treatment. However, it remained unclear whether these two effects were mediated by two different receptor subtypes or by a single receptor that coupled to multiple G proteins.
More recently, several other examples for Ca2+ channel modulation via endogenous P2Y receptors were reported. Vartian and Boehm [91] found that adenosine nucleotides inhibited voltage-gated Ca2+ currents in PC12 cells via a voltage-dependent and PTX-sensitive mechanism, but at that time the pharmacological profile of the receptor involved did not correspond to any of the known P2Y receptors. However, in subsequent studies the receptor responsible for this inhibition of voltage-gated Ca2+ currents in PC12 cells could be identified as the P2Y12 receptor [92, 93]. In hamster submandibular neurons, the P2Y2 receptor was shown to mediate a voltage-dependent and PTX-sensitive inhibition of voltage-gated Ca2+ currents [94]. Moreover, P2Y1 receptors in rat dorsal root ganglion neurons [95] and P2Y12 receptors in rat sympathetic neurons [96] were both shown to mediate a voltage-dependent and pertussis toxin-sensitive inhibition of N-type calcium channels.
However, not only inhibitory effects of nucleotides on Ca2+ channels have been observed. ATP has also been shown to increase calcium currents in rat cardiac cells [97] and in hippocampal neurons [98]. Unfortunately, the signalling cascades involved in these effects have not been elucidated in detail.
Recombinant receptors
Based on their findings that UTP activates pertussis toxin-sensitive as well as -resistant pathways in NG108-15 neuronal hybrid cells [90, 99], Alexander Filippov, Eric Barnard, David Brown and collaborators extended their investigations on the coupling of P2Y receptors to neuronal ion channels by using rat superior cervical ganglion neurons as an expression system for molecularly defined receptor subtypes. All of the P2Y receptors that were investigated (P2Y1, 2, 4, 6, 12) did couple to voltage-gated Ca2+ channels. As expected for a Gi/o coupled receptor, the inhibition of voltage-gated Ca2+ currents by P2Y12 was voltage-dependent and PTX-sensitive, and thus appeared to involve only the membrane-delimited pathway [14]. By contrast, the inhibition of voltage-gated Ca2+ currents mediated by P2Y1, 2, 4 and 6 included two components: (1) the voltage-dependent and PTX-sensitive membrane-delimited pathway as well as a (2) PTX-resistant component. The P2Y2 receptor-mediated inhibition included a voltage-dependent and PTX-sensitive component, on one hand, and a voltage-independent and PTX-resistant component, on the other hand [100]. By contrast, when P2Y6 [101] or P2Y1 [102, 103] were overexpressed, the pertussis toxin-resistant component appeared to be voltage-dependent, since it was facilitated by a large depolarizing prepulse. Interestingly, the authors found that the inhibition mediated by the P2Y6 receptor is much more pronounced in perforated patch (73%) as compared to whole-cell (53%) recordings. Moreover, the inhibition observed in the perforated-patch configuration was hardly altered by PTX, while in whole-cell recordings it was reduced by 60% after treatment with this toxin. Similar results were obtained when P2Y4 receptors were overexpressed [104]. In that case almost no inhibition (∼10%) was observed in the disrupted whole-cell mode, while currents recorded in the perforated-patch mode were reduced by ∼45% in a PTX-sensitive and voltage-dependent manner. Taken together, these findings indicate that even the membrane-delimited, PTX-sensitive pathway may require a soluble co-factor, and that a modulation of voltage-gated Ca2+ currents should rather be investigated in the perforatedpatch than the whole-cell mode of the patch clamp technique. Unfortunately, most of our current knowledge concerning the regulation of neuronal Ca2+ channels via P2Y and other G protein-coupled receptors stems from whole-cell experiments only.
Regulation of K+ channels by P2Y receptors
Native receptors
Besides voltage-gated Ca2+ channels, a variety of K+ channels have consistently been shown to be modulated by nucleotides. One of the first examples was the inhibition of I M by UTP [105] and ATP [106] in bullfrog sympathetic neurons. Although the receptors responsible for these effects remained unknown at that time, the authors could subsequently show that I M inhibition by nucleotides involved a G protein [107, 108]. Only a few years later, after the cloning of the first G protein-coupled nucleotide receptors, Filippov et al. [99] could demonstrate that activation of the PLC-linked P2Y2 receptor in NG108-15 neuroblastoma × glioma hybrid cells led to an inhibition of native I M. Subsequently, several other groups could also observe an inhibition of I M mediated by native P2Y receptors. In rat superior cervical ganglion neurons, a uridine nucleotide preferring receptor, most likely P2Y6, was reported to mediate an inhibition of I M [109]. The signalling cascade underlying this inhibition included activation of PLC, generation of IP3, and release of Ca2+ from intracellular stores [110]. In rat thoracolumbar sympathetic neurons [111], UTP and UDP reduced I M in cultures isolated from 9- to 12-day-old rats, but not in cultures prepared from newborn animals. More recently, Meng et al. [112] could show that the nucleotide induced inhibition of I M in bullfrog sympathetic neurons is mediated by a P2Y receptor, most likely P2Y4, as suggested by the observed rank order of agonist potencies.
In addition to I M modulation, several other neuronal potassium channels were also found to be modulated by nucleotides. In a series of papers published in the mid nineties, Ikeuchi and Nishizaki described an outwardly rectifying potassium current activated by nucleotides. The current was found in various regions of the rat brain, including striatal neurons [113], inferior colliculus neurons [114], superior colliculus neurons [115], cerebellar neurons [116] and in hippocampal neurons [117]. Interestingly, although the current that was induced seemed to be the same in all these studies, the signal transduction mechanisms, as well as the receptor subtypes involved in channel activation seemed to differ between the various regions of the brain that were investigated. Responses that were induced by ATP in striatal neurons and by adenosine in superior colliculus and hippocampal neurons involved a diffusible second messenger and protein kinase C, as could be shown by single channel recordings from cell-attached patches and by using the protein kinase C inhibitor GF109203X, respectively. By contrast, the actions of ADP in inferior colliculus neurons were membrane delimited, presumably based on a direct interaction of βγ subunits of a PTX-insensitive G protein with the channel protein, as could be shown by single channel recordings from outside-out patches.
In rat hippocampal neurons, ATP was also reported to inhibit a voltage-gated K+ channel [118]. UTP was as potent as ATP, and ADP and α,β-methylene ATP also inhibited the outward current. More recently, another voltage-gated potassium channel, a fast delayed inward rectifier, in Xenopus spinal neurons was found to be inhibited by adenine and uridine nucleotides [119]. Since a much larger proportion of the neurons responded to ADP, but not to ATP or UTP, the authors speculated about the presence of two different P2Y receptor subtypes, one mediating the effects of the triphosphates and the other mediating the effects of ADP.
Recombinant receptors
The potassium channels that have been studied most extensively with respect to their modulation by heterologously expressed P2Y receptors are the KCNQ channels and the GIRKs. Among the receptors that were investigated, the Gq/11 coupled receptors P2Y1,2,4 and P2Y6, when expressed in rat superior cervical ganglion neurons, all inhibited the I M in a PTX-resistant manner [100, 101, 103, 104]. However, when the P2Y12 receptor was expressed in these neurons, no inhibition of I M by nucleotides could be observed [14]. When rat GIRK1 and GIRK2 (Kir3.1 and 3.2 subunits) were co-expressed, 2MeSADP and 2MeSATP, two potent P2Y12 receptor agonists, evoked a K+ current through GIRK channels. This is in accordance with the general concept that Gi/o coupled receptors mediate an activation of GIRK channels. A more unexpected finding was that the Gq/11-linked P2Y1 receptor, when co-expressed with Kir3.1 and 3.2 subunits instead of P2Y12, also produced a PTX-sensitive activation of GIRK. This further supports the idea that a single P2Y receptor may couple to multiple G proteins. However, with P2Y1, the fast activation was followed by a slower, but almost complete inactivation of the current in the continued presence of the agonist, an effect that has previously been observed when rat P2Y2 receptors were expressed together with GIRK1/4 in Xenopus oocytes [120]. Similar results were also obtained with the mouse P2Y2 receptor, but not with human P2Y6, when expressed together with GIRK channels in Xenopus oocytes [121].
Regulation of ligand-gated ion channels by P2Y receptors
Native receptors
In neurons, nucleotides can act not only on voltage-gated, but also on ligand-gated ion channels. In layer V pyramidal neurons of the rat prefrontal cortex, adenine and uridine nucleotides, presumably acting on P2Y2 receptors, were found to enhance currents elicited by N-methlyl-d-aspartate (NMDA) [122]. By contrast, ADP-βS, a potent P2Y1 receptor agonist, inhibited currents through NMDA receptors [123] in the same cells. Since this inhibition was antagonized by PPADS and MRS2179, an involvement of the P2Y1 receptor seemed most likely. In addition, ATP was found to directly inhibit NMDA receptors independently of G proteins and, therefore, not by activation of P2Y receptors. This latter inhibition was revealed to involve a direct binding of the nucleotide to the glutamate-binding pocket of the NR2B subunit of NMDA receptors [124].
Another ligand-gated ion channel that was found to be modulated by nucleotides is the vanilloid receptor 1 (VR1). In rat dorsal root ganglion neurons, capsaicin-evoked currents through VR1 were enhanced by the application of nucleotides [125]. This nucleotide-induced potentiation was abolished by application of calphostin C, a potent protein kinase C inhibitor, and was mimicked by phorbol esters, thus suggesting that a P2Y receptor linked to protein kinase C via Gq/11 proteins was involved. Considering the observed rank order of agonist potencies, the P2Y1 receptor was considered to be the most likely candidate. However, more recently the same group could show that the ATP-induced potentiation can also be observed in dorsal root ganglion neurons of P2Y1-deficient mice [126]. Moreover, in in situ hybridizations rat dorsal root ganglion neurons were found to co-express VR1 and P2Y2 mRNA, but not P2Y1 mRNA. UTP was reported to be as potent an agonist as ATP, and suramin (which blocks P2Y2 but not P2Y4) abolished the potentiation by UTP. Taken together, these results indicated that it was rather the P2Y2 than the P2Y1 receptor that mediated the effects of nucleotides on VR1 in mouse and rat DRG neurons.
Recombinant receptors
In addition to the above effects obtained with native P2Y receptors in primary neuronal cell cultures, akin results were also obtained when P2Y receptors were heterologously expressed in Xenopus oocytes. When co-expressed together with the P2X1 receptor, activation of both, P2Y1 and P2Y2, caused a significant potentiation of the α,β-MeATP-induced current through P2X1 [127].
Functional consequences of neuronal ion channel regulation via P2Y receptors
The effects of P2Y receptor activation on neuronal ion channels that have been observed most consistently and frequently are (1) inhibition of voltage-activated Ca2+ currents, (2) activation of Ca2+-dependent K+ channels or GIRKs, and (3) inhibition of I M. Voltage-gated Ca2+ channels, in particular N- and P/Q-type channels, are located at presynaptic nerve terminals and link invading action potentials to transmembrane Ca2+ influx and ensuing vesicle exocytosis. Accordingly, the modulation of these ion channels via G protein-coupled receptors will lead to changes in transmitter release [128]. The kinetics of presynaptic action potentials are shaped by voltage-activated and Ca2+-dependent K+ channels, and modulation of these channels via G protein-coupled receptors can also lead to changes in vesicle exocytosis [129, 130]. In contrast to these K+ channels, inwardly rectifying K+ channels including GIRKs are hardly found at presynaptic nerve terminals and are generally not believed to directly contribute to transmitter release [129, 131]. With respect to KCNQ channels, which mediate I M, contrasting results have been obtained. In peripheral sympathetic neurons, no evidence could be obtained for a role of these ion channels in presynaptic transmitter release [132, 133]. However, in central synaptosomes, modulators of these ion channels were found to affect the release of various transmitters [134]. Hence, the regulation of Ca2+ channels and Ca2+-dependent K+ channels via P2Y receptors will preferentially lead to changes in synaptic transmission via a presynaptic modulation of transmitter release, whereas the control of GIRKs and KCNQ channels will rather cause changes in the postsynaptic excitability. The modulation of ligand-gated ion channels may lead to pre- as well as postsynaptic effects depending on where the regulated ionotropic receptors are located [135]. In fact, ATP and other nucleotides acting at P2Y receptors may cause both, pre- and postsynaptic effects in the central as well as peripheral nervous system.
In the cortex and hippocampus, nucleotides were found to inhibit the release of acetylcholine and noradrenaline via presynaptic sites of action. With respect to the release of dopamine, serotonin and glutamate, inhibitory as well as facilitatory presynaptic effects have been described for nucleotides in a variety of brain regions. The release of the inhibitory transmitters GABA and glycine, however, is rather enhanced than reduced by nucleotides. The precise receptor subtypes involved in these presynaptic effects remained mostly unknown, and only the facilitatory effects were evidenced to be mediated by P2X receptors. Moreover, a clear-cut role of presynaptic Ca2+ or K+ channels in the modulatory effects of nucleotides was not established, as reviewed by Cunha and Ribeiro [136]. In hippocampal neurons, a presynaptic P2Y receptor mediated an ATP-dependent inhibition of excitatory, but not of inhibitory, synaptic transmission via PTX-sensitive G proteins, and an inhibition of voltage-activated Ca2+ currents. However, the receptor subtype(s) involved were not further characterized [137]. In the medial habenula, a presynaptic P2Y4-like receptor was shown to enhance glutamate release, whereas a presumed P2Y2-like receptor mediated an inhibition [138]. Whether these presynaptic P2Y receptors affected transmitter release through a modulation of Ca2+ or K+ channels remained unfortunately unknown. In the prefrontal and parietal cortex, activation of a P2Y1-like receptor reduced glutamatergic transmission, but only the component involving NMDA receptors. Furthermore, the receptor mediated an inhibition of depolarizations caused by N-methlyl-d-aspartate, but not of those induced by alpha-amino-3- hydroxy-5-methyl-4-isoxazole propionic acid [123]. Thus, P2Y receptor activation may also interfere with synaptic transmission via a modulation of postsynaptic ionotropic receptors.
In the periphery, in autonomic and motor neurons, release of acteylcholine or noradrenaline was also reported to be controlled by presynaptic P2X and P2Y receptors [136]. The system that has been studied in greatest detail is the sympathetic nervous system, since ATP and noradrenaline have long been known as cotransmitters there. Early evidence for presynaptic nucleotide receptors in sympathetic neurons has been obtained 20 years ago [139]. More recently, several nucleotides were found to reduce, whereas P2 receptor antagonists were found to enhance, noradrenaline release from the mouse vas deferens as indication of inhibitory presynaptic P2Y autoreceptors [140]. In numerous other sympathetically innervated tissues, nucleotides were also reported to inhibit transmitter release [141, 142]. For a long period of time, the P2Y receptor subtypes involved in these effects remained unknown, but recent evidence suggests that P2Y12 and/or P2Y13 receptors mediate autoinhibition of transmitter release from sympathetic neurons [143]. In PC12 cells which are ontogenetically related to sympathetic neurons, P2Y12 receptors mediate an inhibition of voltage-activated Ca2+ channels (see above and [91–93]). Most recently, this effect was also observed in primary cultures of rat superior cervical ganglion neurons and it was demonstrated to mediate autoinhibition of transmitter release [96]. Hence, sympatho-effector transmission is regulated by an inhibition of voltage-activated Ca2+ channels via presynaptic P2Y12 receptors. In sensory neurons, in contrast, P2Y1 receptors mediate an inhibition of voltage-activated Ca2+ channels [144], and this effect apparently leads to reduced synaptic transmission in pain pathways and to analgesic effects [95].
However, P2Y receptor activation mediates not only inhibitory, but also excitatory effects in sensory neurons. P2Y1 receptor activation, in particular, was suggested to facilitate touch-induced impulse generation in sensory neurons [145], but the underlying mechanisms remained unknown. In this context, the interaction with VR1 receptors might be a relevant mechanism (see above). In autonomic neurons, P2Y1 receptors were also involved in excitatory postsynaptic effects: Excitatory postsynaptic potentials that were blocked by a P2Y1 receptor antagonist were observed in the enteric nervous system. Direct activation of these receptors caused a Na+-dependent increase in membrane conductance, but the precise ionic mechanisms were not further investigated [146]. In postganglionic sympathetic neurons, P2Y receptors were also found to mediate excitatory postsynaptic effects. UTP depolarizes sympathetic ganglia and triggers transmitter release from dissociated sympathetic neurons via receptors different from those activated by ATP [147, 148]. The secretagogue action of UTP was mimicked equipotently by UDP [111, 148], and both nucleotides also caused an inhibition of I M [109, 111]. The receptor involved was evidenced to be P2Y6. However, the stimulatory effect did not only involve the inhibition of KCNQ channels, but also an activation of protein kinase C [149].
Conclusion
The modulation of neuronal ion channels via G protein-coupled receptors is a major means of adapting the responsiveness of a neuron to changes in environmental conditions [1]. Such modulatory effects have been described for virtually all known neurotransmitters and nucleotides are no exception. Within the last 15 years, 8 different G protein-coupled nucleotide receptors have been characterized, and numerous examples of ion channels that are regulated via one or more of these receptors are described here. Thus, aside of their functions as fast synaptic transmitters acting at P2X receptors [150], nucleotides also fulfil roles as neuromodulators, as do many other classical transmitter, such as glutamate, GABA, acetylcholine, and serotonin. Thus, this summary of ion channels regulated via P2Y receptors adds ATP and related nucleotides to this list of pluripotent neurotransmitters.
References
L Kaczmarek IB Levitan (1987) Neuromodulation: The Biochemical Control of Neuronal Excitability Oxford University Press New York
CM Anderson FE Parkinson (1997) ArticleTitlePotential signalling roles for UTP and UDP: Sources, regulation and release of uracil nucleotides Trends Pharmacol Sci 18 IssueID10 387–392 Occurrence Handle9357323 Occurrence Handle10.1016/S0165-6147(97)01106-1 Occurrence Handle1:CAS:528:DyaK2sXmvFegt7w%3D
ER Lazarowski RC Boucher TK Harden (2003) ArticleTitleMechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules Mol Pharmacol 64 IssueID4 785–795 Occurrence Handle14500734 Occurrence Handle10.1124/mol.64.4.785 Occurrence Handle1:CAS:528:DC%2BD3sXnslGrtbk%3D
V Ralevic G Burnstock (1998) ArticleTitleReceptors for purines and pyrimidines Pharmacol Rev 50 IssueID3 413–492 Occurrence Handle9755289 Occurrence Handle1:CAS:528:DyaK1cXmvFamur0%3D
RA North (2002) ArticleTitleMolecular physiology of P2X receptors Physiol Rev 82 IssueID4 1013–1067 Occurrence Handle12270951 Occurrence Handle1:CAS:528:DC%2BD38Xot1Ggu78%3D
KA Jacobson MF Jarvis M Williams (2002) ArticleTitlePurine and pyrimidine (P2) receptors as drug targets J Med Chem 45 IssueID19 4057–4093 Occurrence Handle12213051 Occurrence Handle10.1021/jm020046y Occurrence Handle1:CAS:528:DC%2BD38Xmt1Gitr0%3D
K Sak TE Webb (2002) ArticleTitleA retrospective of recombinant P2Y receptor subtypes and their pharmacology Arch Biochem Biophys 397 IssueID1 131–136 Occurrence Handle11747319 Occurrence Handle10.1006/abbi.2001.2616 Occurrence Handle1:CAS:528:DC%2BD3MXptFWrsbc%3D
MP Abbracchio JM Boeynaems EA Barnard et al. (2003) ArticleTitleCharacterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family Trends Pharmacol Sci 24 IssueID2 52–55 Occurrence Handle12559763 Occurrence Handle10.1016/S0165-6147(02)00038-X Occurrence Handle1:CAS:528:DC%2BD3sXmt1altg%3D%3D
H Inbe S Watanabe M Miyawaki et al. (2004) ArticleTitleIdentification and characterization of a cell-surface receptor, P2Y15, for AMP and adenosine J Biol Chem 279 IssueID19 19790–19799 Occurrence Handle15001573 Occurrence Handle10.1074/jbc.M400360200 Occurrence Handle1:CAS:528:DC%2BD2cXjs1Wit7c%3D
W He FJ Miao DC Lin et al. (2004) ArticleTitleCitric acid cycle intermediates as ligands for orphan G-protein-coupled receptors Nature 429 IssueID6988 188–193 Occurrence Handle15141213 Occurrence Handle10.1038/nature02488 Occurrence Handle1:CAS:528:DC%2BD2cXjvVKgsLs%3D
C Kennedy AD Qi CL Herold et al. (2000) ArticleTitleATP, an agonist at the rat P2Y(4) receptor, is an antagonist at the human P2Y(4) receptor Mol Pharmacol 57 IssueID5 926–931 Occurrence Handle10779375 Occurrence Handle1:CAS:528:DC%2BD3cXjtVegur0%3D
PJ White TE Webb MR Boarder (2003) ArticleTitleCharacterization of a Ca2+ response to both UTP and ATP at human P2Y11 receptors: Evidence for agonist-specific signaling Mol Pharmacol 63 IssueID6 1356–1363 Occurrence Handle12761346 Occurrence Handle10.1124/mol.63.6.1356 Occurrence Handle1:CAS:528:DC%2BD3sXkt1SgsrY%3D
AD Qi AC Zambon PA Insel RA Nicholas (2001) ArticleTitleAn arginine/glutamine difference at the juxtaposition of transmembrane domain 6 and the third extracellular loop contributes to the markedly different nucleotide selectivities of human and canine P2Y11 receptors Mol Pharmacol 60 IssueID6 1375–1382 Occurrence Handle11723245 Occurrence Handle1:CAS:528:DC%2BD3MXovVOitLg%3D
J Simon AK Filippov S Goransson et al. (2002) ArticleTitleCharacterization and channel coupling of the P2Y(12) nucleotide receptor of brain capillary endothelial cells J Biol Chem 277 IssueID35 31390–31400 Occurrence Handle12080041 Occurrence Handle10.1074/jbc.M110714200 Occurrence Handle1:CAS:528:DC%2BD38XmslOqsL4%3D
ET Bodor GL Waldo SB Hooks et al. (2003) ArticleTitlePurification and functional reconstitution of the human P2Y12 receptor Mol Pharmacol 64 IssueID5 1210–1216 Occurrence Handle14573771 Occurrence Handle10.1124/mol.64.5.1210 Occurrence Handle1:CAS:528:DC%2BD3sXos1Snsb8%3D
FL Zhang L Luo E Gustafson et al. (2002) ArticleTitleP2Y(13): Identification and characterization of a novel Galphai-coupled ADP receptor from human and mouse J Pharmacol Exp Ther 301 IssueID2 705–713 Occurrence Handle11961076 Occurrence Handle10.1124/jpet.301.2.705 Occurrence Handle1:CAS:528:DC%2BD38XjtlKis7s%3D
F Marteau E Poul ParticleLe D Communi et al. (2003) ArticleTitlePharmacological characterization of the human P2Y13 receptor Mol Pharmacol 64 IssueID1 104–112 Occurrence Handle12815166 Occurrence Handle10.1124/mol.64.1.104 Occurrence Handle1:CAS:528:DC%2BD3sXkvV2qtrY%3D
M Fumagalli L Trincavelli D Lecca et al. (2004) ArticleTitleCloning, pharmacological characterisation and distribution of the rat G-protein-coupled P2Y(13) receptor Biochem Pharmacol 68 IssueID1 113–124 Occurrence Handle15183123 Occurrence Handle10.1016/j.bcp.2004.02.038 Occurrence Handle1:CAS:528:DC%2BD2cXkslWksr4%3D
JK Chambers LE Macdonald HM Sarau et al. (2000) ArticleTitleA G protein-coupled receptor for UDP-glucose J Biol Chem 275 IssueID15 10767–10771 Occurrence Handle10753868 Occurrence Handle10.1074/jbc.275.15.10767 Occurrence Handle1:CAS:528:DC%2BD3cXislShsrg%3D
RA Nicholas WC Watt ER Lazarowski et al. (1996) ArticleTitleUridine nucleotide selectivity of three phospholipase C-activating P2 receptors: Identification of a UDP-selective, a UTP-selective, and an ATP- and UTP-specific receptor Mol Pharmacol 50 IssueID2 224–229 Occurrence Handle8700127 Occurrence Handle1:CAS:528:DyaK28XkvFCqt7Y%3D
RW Peoples C Li (1998) ArticleTitleInhibition of NMDA-gated ion channels by the P2 purinoceptor antagonists suramin and reactive blue 2 in mouse hippocampal neurones Br J Pharmacol 124 IssueID2 400–408 Occurrence Handle9641559 Occurrence Handle10.1038/sj.bjp.0701842 Occurrence Handle1:CAS:528:DyaK1cXjs1Ogurg%3D
LJ Galietta S Falzoni F Virgilio ParticleDi et al. (1997) ArticleTitleCharacterization of volume-sensitive taurine- and Cl(−)-permeable channels Am J Physiol 273 IssueID1 Pt 1 C57–C66 Occurrence Handle9252442 Occurrence Handle1:CAS:528:DyaK2sXlt1Skuro%3D
JL Boyer T Romero-Avila JB Schachter TK Harden (1996) ArticleTitleIdentification of competitive antagonists of the P2Y1 receptor Mol Pharmacol 50 IssueID5 1323–1329 Occurrence Handle8913364 Occurrence Handle1:CAS:528:DyaK28XntFCqsLk%3D
E Nandanan E Camaioni SY Jang et al. (1999) ArticleTitleStructure–activity relationships of bisphosphate nucleotide derivatives as P2Y1 receptor antagonists and partial agonists J Med Chem 42 IssueID9 1625–1638 Occurrence Handle10229631 Occurrence Handle10.1021/jm980657j Occurrence Handle1:CAS:528:DyaK1MXisVGhu74%3D
G Hollopeter HM Jantzen D Vincent et al. (2001) ArticleTitleIdentification of the platelet ADP receptor targeted by antithrombotic drugs Nature 409 IssueID6817 202–207 Occurrence Handle11196645 Occurrence Handle10.1038/35051599 Occurrence Handle1:CAS:528:DC%2BD3MXlvFSktQ%3D%3D
F Storey (2001) ArticleTitleThe P2Y12 receptor as a therapeutic target in cardiovascular disease Platelets 12 IssueID4 197–209 Occurrence Handle11454254 Occurrence Handle10.1080/09537100120058739 Occurrence Handle1:CAS:528:DC%2BD3MXkvFWhu7Y%3D
LK Mamedova BV Joshi ZG Gao et al. (2004) ArticleTitleDiisothiocyanate derivatives as potent, insurmountable antagonists of P2Y6 nucleotide receptors Biochem Pharmacol 67 IssueID9 1763–1770 Occurrence Handle15081875 Occurrence Handle10.1016/j.bcp.2004.01.011 Occurrence Handle1:CAS:528:DC%2BD2cXjtVemtr8%3D
D Communi NS Gonzalez M Detheux et al. (2001) ArticleTitleIdentification of a novel human ADP receptor coupled to G(i) J Biol Chem 276 IssueID44 41479–41485 Occurrence Handle11546776 Occurrence Handle10.1074/jbc.M105912200 Occurrence Handle1:CAS:528:DC%2BD3MXosVKgtbo%3D
D Moore J Chambers H Waldvogel et al. (2000) ArticleTitleRegional and cellular distribution of the P2Y(1) purinergic receptor in the human brain: Striking neuronal localisation J Comp Neurol 421 IssueID3 374–384 Occurrence Handle10813793 Occurrence Handle10.1002/(SICI)1096-9861(20000605)421:3<374::AID-CNE6>3.0.CO;2-Z Occurrence Handle1:CAS:528:DC%2BD3cXjtlKlt7g%3D
DJ Moore JK Chambers JP Wahlin et al. (2001) ArticleTitleExpression pattern of human P2Y receptor subtypes: A quantitative reverse transcription-polymerase chain reaction study Biochim Biophys Acta 1521 IssueID1–3 107–119 Occurrence Handle11690642 Occurrence Handle1:CAS:528:DC%2BD3MXot1Sju7c%3D
MJ Moran-Jimenez C Matute (2000) ArticleTitleImmunohistochemical localization of the P2Y(1) purinergic receptor in neurons and glial cells of the central nervous system Brain Res Mol Brain Res 78 IssueID1–2 50–58 Occurrence Handle10891584 Occurrence Handle10.1016/S0169-328X(00)00067-X Occurrence Handle1:CAS:528:DC%2BD3cXks12qsrs%3D
FL Zhang L Luo E Gustafson et al. (2001) ArticleTitleADP is the cognate ligand for the orphan G protein-coupled receptor SP1999 J Biol Chem 276 IssueID11 8608–8615 Occurrence Handle11104774 Occurrence Handle10.1074/jbc.M009718200 Occurrence Handle1:CAS:528:DC%2BD3MXitFKgtbc%3D
D Communi C Govaerts M Parmentier JM Boeynaems (1997) ArticleTitleCloning of a human purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase J Biol Chem 272 IssueID51 31969–31973 Occurrence Handle9405388 Occurrence Handle10.1074/jbc.272.51.31969 Occurrence Handle1:CAS:528:DyaK1cXislCgtg%3D%3D
AR Cantrell WA Catterall (2001) ArticleTitleNeuromodulation of Na+ channels: An unexpected form of cellular plasticity Nat Rev Neurosci 2 IssueID6 397–407 Occurrence Handle11389473 Occurrence Handle10.1038/35077553 Occurrence Handle1:CAS:528:DC%2BD3MXksFOltbo%3D
AR Cantrell RD Smith AL Goldin et al. (1997) ArticleTitleDopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel alpha subunit J Neurosci 17 IssueID19 7330–7338 Occurrence Handle9295379 Occurrence Handle1:CAS:528:DyaK2sXmtl2lsbY%3D
AR Cantrell T Scheuer WA Catterall (1999) ArticleTitleVoltage-dependent neuromodulation of Na+ channels by D1-like dopamine receptors in rat hippocampal neurons J Neurosci 19 IssueID13 5301–5310 Occurrence Handle10377341 Occurrence Handle1:CAS:528:DyaK1MXktlOqur4%3D
AR Cantrell JY Ma T Scheuer WA Catterall (1996) ArticleTitleMuscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons Neuron 16 IssueID5 1019–1026 Occurrence Handle8630240 Occurrence Handle10.1016/S0896-6273(00)80125-7 Occurrence Handle1:CAS:528:DyaK28XjsVKgsLo%3D
T Mittmann C Alzheimer (1998) ArticleTitleMuscarinic inhibition of persistent Na+ current in rat neocortical pyramidal neurons J Neurophysiol 79 IssueID3 1579–1582 Occurrence Handle9497434 Occurrence Handle1:CAS:528:DyaK1cXitlChtr8%3D
DB Carr SR Sesack (1996) ArticleTitleHippocampal afferents to the rat prefrontal cortex: Synaptic targets and relation to dopamine terminals J Comp Neurol 369 IssueID1 1–15 Occurrence Handle8723699 Occurrence Handle10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7 Occurrence Handle1:CAS:528:DyaK28XjtlSgsbk%3D
MS Gold DB Reichling MJ Shuster JD Levine (1996) ArticleTitleHyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors Proc Natl Acad Sci USA 93 IssueID3 1108–1112 Occurrence Handle8577723 Occurrence Handle10.1073/pnas.93.3.1108 Occurrence Handle1:CAS:528:DyaK28Xptl2lsg%3D%3D
S England S Bevan RJ Docherty (1996) ArticleTitlePGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade J Physiol 495 IssueIDPt 2 429–440 Occurrence Handle8887754 Occurrence Handle1:CAS:528:DyaK28XmtVylu70%3D
WA Catterall J Striessnig TP Snutch E Perez-Reyes (2003) ArticleTitleInternational Union of Pharmacology: XL. Compendium of voltage-gated ion channels: Calcium channels Pharmacol Rev 55 IssueID4 579–581 Occurrence Handle14657414 Occurrence Handle10.1124/pr.55.4.8 Occurrence Handle1:CAS:528:DC%2BD2cXhtlWktw%3D%3D
B Hille (1994) ArticleTitleModulation of ion-channel function by G-protein-coupled receptors Trends Neurosci 17 IssueID12 531–536 Occurrence Handle7532338 Occurrence Handle10.1016/0166-2236(94)90157-0 Occurrence Handle1:CAS:528:DyaK2MXit1yqs74%3D
SR Ikeda K Dunlap (1999) ArticleTitleVoltage-dependent modulation of N-type calcium channels: Role of G protein subunits Adv Second Messenger Phosphoprot Res 33 131–151 Occurrence Handle1:STN:280:DyaK1M3jsVOnsg%3D%3D
AC Dolphin (1999) ArticleTitleL-type calcium channel modulation Adv Second Messenger Phosphoprot Res 33 153–177 Occurrence Handle1:STN:280:DyaK1M3jsVOnsw%3D%3D
AC Dolphin (2003) ArticleTitleG protein modulation of voltage-gated calcium channels Pharmacol Rev 55 IssueID4 607–627 Occurrence Handle14657419 Occurrence Handle10.1124/pr.55.4.3 Occurrence Handle1:CAS:528:DC%2BD2cXhtlWrsg%3D%3D
M Diverse-Pierluissi K Dunlap (1993) ArticleTitleDistinct, convergent second messenger pathways modulate neuronal calcium currents Neuron 10 IssueID4 753–760 Occurrence Handle8097399 Occurrence Handle10.1016/0896-6273(93)90175-Q Occurrence Handle1:CAS:528:DyaK3sXmsVWksbk%3D
S Boehm S Huck M Freissmuth (1996) ArticleTitleInvolvement of a phorbol ester-insensitive protein kinase C in the alpha2-adrenergic inhibition of voltage-gated calcium current in chick sympathetic neurons J Neurosci 16 IssueID15 4596–4603 Occurrence Handle8764648 Occurrence Handle1:CAS:528:DyaK28XksFWjurg%3D
PJ Kammermeier V Ruiz-Velasco SR Ikeda (2000) ArticleTitleA voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Galpha q/11 and Gbeta gamma J Neurosci 20 IssueID15 5623–5629 Occurrence Handle10908599 Occurrence Handle1:CAS:528:DC%2BD3cXlt1GltLg%3D
JE Haley P Delmas S Offermanns et al. (2000) ArticleTitleMuscarinic inhibition of calcium current and M current in Galpha q-deficient mice J Neurosci 20 IssueID11 3973–3979 Occurrence Handle10818132 Occurrence Handle1:CAS:528:DC%2BD3cXjsFeltLY%3D
GA Gutman KG Chandy JP Adelman et al. (2003) ArticleTitleInternational Union of Pharmacology: XLI. Compendium of voltage-gated ion channels: Potassium channels Pharmacol Rev 55 IssueID4 583–586 Occurrence Handle14657415 Occurrence Handle10.1124/pr.55.4.9 Occurrence Handle1:CAS:528:DC%2BD2cXhtlWktA%3D%3D
PR Stanfield S Nakajima Y Nakajima (2002) ArticleTitleConstitutively active and G-protein coupled inward rectifier K+ channels: Kir2.0 and Kir3.0 Rev Physiol Biochem Pharmacol 145 47–179 Occurrence Handle12224528 Occurrence Handle10.1007/BFb0116431 Occurrence Handle1:CAS:528:DC%2BD38XotVSjtLk%3D
S Peleg D Varon T Ivanina et al. (2002) ArticleTitleG(alpha)(i) controls the gating of the G protein-activated K(+) channel, GIRK Neuron 33 IssueID1 87–99 Occurrence Handle11779482 Occurrence Handle10.1016/S0896-6273(01)00567-0 Occurrence Handle1:CAS:528:DC%2BD38XksFSitA%3D%3D
A Benians JL Leaney G Milligan A Tinker (2003) ArticleTitleThe dynamics of formation and action of the ternary complex revealed in living cells using a G-protein-gated K+ channel as a biosensor J Biol Chem 278 IssueID12 10851–10858 Occurrence Handle12529316 Occurrence Handle10.1074/jbc.M212299200 Occurrence Handle1:CAS:528:DC%2BD3sXitVCqurw%3D
CA Doupnik N Davidson HA Lester P Kofuji (1997) ArticleTitleRGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels Proc Natl Acad Sci USA 94 IssueID19 10461–10466 Occurrence Handle9294233 Occurrence Handle10.1073/pnas.94.19.10461 Occurrence Handle1:CAS:528:DyaK2sXmt1Gjt7o%3D
CL Huang S Feng DW Hilgemann (1998) ArticleTitleDirect activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma Nature 391 IssueID6669 803–806 Occurrence Handle9486652 Occurrence Handle10.1038/35882 Occurrence Handle1:CAS:528:DyaK1cXhtlCjurc%3D
MJ Rebecchi SN Pentyala (2000) ArticleTitleStructure, function, and control of phosphoinositide-specific phospholipase C Physiol Rev 80 IssueID4 1291–1335 Occurrence Handle11015615 Occurrence Handle1:CAS:528:DC%2BD3cXnsVKisL0%3D
MD Mark S Herlitze (2000) ArticleTitleG-protein mediated gating of inward-rectifier K+ channels Eur J Biochem 267 IssueID19 5830–5836 Occurrence Handle10998041 Occurrence Handle10.1046/j.1432-1327.2000.01670.x Occurrence Handle1:CAS:528:DC%2BD3cXnt1Snu7s%3D
C Mullner D Vorobiov AK Bera et al. (2000) ArticleTitleHeterologous facilitation of G protein-activated K(+) channels by beta-adrenergic stimulation via cAMP-dependent protein kinase J Gen Physiol 115 IssueID5 547–558 Occurrence Handle10779313 Occurrence Handle10.1085/jgp.115.5.547 Occurrence Handle1:CAS:528:DC%2BD3cXjtl2hu74%3D
J Robbins (2001) ArticleTitleKCNQ potassium channels: Physiology, pathophysiology, and pharmacology Pharmacol Ther 90 IssueID1 1–19 Occurrence Handle11448722 Occurrence Handle10.1016/S0163-7258(01)00116-4 Occurrence Handle1:CAS:528:DC%2BD3MXltVCqt7g%3D
DA Brown (1983) ArticleTitleSlow cholinergic excitation—a mechanism for increasing neuronal excitability TINS 6 302–307
NV Marrion (1997) ArticleTitleControl of M-current Annu Rev Physiol 59 483–504 Occurrence Handle9074774 Occurrence Handle10.1146/annurev.physiol.59.1.483 Occurrence Handle1:CAS:528:DyaK2sXhvVSjtb0%3D
JE Haley FC Abogadie P Delmas et al. (1998) ArticleTitleThe alpha subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons J Neurosci 18 IssueID12 4521–4531 Occurrence Handle9614229 Occurrence Handle1:CAS:528:DyaK1cXjslGjsb0%3D
BC Suh B Hille (2002) ArticleTitleRecovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis Neuron 35 IssueID3 507–520 Occurrence Handle12165472 Occurrence Handle10.1016/S0896-6273(02)00790-0 Occurrence Handle1:CAS:528:DC%2BD38Xmt12is78%3D
S Jones DA Brown G Milligan et al. (1995) ArticleTitleBradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11 Neuron 14 IssueID2 399–405 Occurrence Handle7857647 Occurrence Handle10.1016/0896-6273(95)90295-3 Occurrence Handle1:CAS:528:DyaK2MXjvFejsL8%3D
JE Haley FC Abogadie JM Fernandez-Fernandez et al. (2000) ArticleTitleBradykinin, but not muscarinic, inhibition of M-current in rat sympathetic ganglion neurons involves phospholipase C-beta 4 J Neurosci 20 IssueID21 RC105 Occurrence Handle11050147 Occurrence Handle1:STN:280:DC%2BD3crgt1Citg%3D%3D
H Cruzblanca DS Koh B Hille (1998) ArticleTitleBradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons Proc Natl Acad Sci USA 95 IssueID12 7151–7156 Occurrence Handle9618554 Occurrence Handle10.1073/pnas.95.12.7151 Occurrence Handle1:CAS:528:DyaK1cXjslymtro%3D
AA Selyanko DA Brown (1996) ArticleTitleIntracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons Neuron 16 IssueID1 151–162 Occurrence Handle8562079 Occurrence Handle10.1016/S0896-6273(00)80032-X Occurrence Handle1:CAS:528:DyaK28XosFCqsg%3D%3D
LW Role (1984) ArticleTitleSubstance P modulation of acetylcholine-induced currents in embryonic chicken sympathetic and ciliary ganglion neurons Proc Natl Acad Sci USA 81 IssueID9 2924–2928 Occurrence Handle6201867 Occurrence Handle10.1073/pnas.81.9.2924 Occurrence Handle1:CAS:528:DyaL2cXktVCgt7c%3D
DC Valenta JE Downing LW Role (1993) ArticleTitlePeptide modulation of ACh receptor desensitization controls neurotransmitter release from chicken sympathetic neurons J Neurophysiol 69 IssueID3 928–942 Occurrence Handle7681868 Occurrence Handle1:CAS:528:DyaK3sXksVWmsrs%3D
W Tan C Du SA Siegelbaum LW Role (1998) ArticleTitleModulation of nicotinic AChR channels by prostaglandin E2 in chick sympathetic ganglion neurons J Neurophysiol 79 IssueID2 870–878 Occurrence Handle9463448 Occurrence Handle1:CAS:528:DyaK1cXhsVGru7k%3D
DM Liu J Cuevas DJ Adams (2000) ArticleTitleVIP and PACAP potentiation of nicotinic ACh-evoked currents in rat parasympathetic neurons is mediated by G-protein activation Eur J Neurosci 12 IssueID7 2243–2251 Occurrence Handle10947803 Occurrence Handle10.1046/j.1460-9568.2000.00116.x Occurrence Handle1:STN:280:DC%2BD3cvjtlenug%3D%3D
W Norenberg M Bek N Limberger et al. (1995) ArticleTitleInhibition of nicotinic acetylcholine receptor channels in bovine adrenal chromaffin cells by Y3-type neuropeptide Y receptors via the adenylate cyclase/protein kinase A system Naunyn-Schmiedeberg’s Arch Pharmacol 351 IssueID4 337–347 Occurrence Handle1:STN:280:DyaK2Mzls1ynsg%3D%3D
GE Yevenes RW Peoples JC Tapia et al. (2003) ArticleTitleModulation of glycine-activated ion channel function by G-protein betagamma subunits Nat Neurosci 6 IssueID8 819–824 Occurrence Handle12858180 Occurrence Handle10.1038/nn1095 Occurrence Handle1:CAS:528:DC%2BD3sXlslOis74%3D
S Ahmadi S Lippross WL Neuhuber HU Zeilhofer (2002) ArticleTitlePGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons Nat Neurosci 5 IssueID1 34–40 Occurrence Handle11740501 Occurrence Handle10.1038/nn778 Occurrence Handle1:CAS:528:DC%2BD38XjtlCquw%3D%3D
WY Lu ZG Xiong S Lei et al. (1999) ArticleTitleG-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors Nat Neurosci 2 IssueID4 331–338 Occurrence Handle10204539 Occurrence Handle10.1038/7243 Occurrence Handle1:CAS:528:DyaK1MXitFGjs78%3D
SA Kotecha JN Oak MF Jackson et al. (2002) ArticleTitleA D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission Neuron 35 IssueID6 1111–1122 Occurrence Handle12354400 Occurrence Handle10.1016/S0896-6273(02)00859-0 Occurrence Handle1:CAS:528:DC%2BD38XnsV2htrw%3D
FJ Lee S Xue L Pei et al. (2002) ArticleTitleDual regulation of NMDA receptor functions by direct protein–protein interactions with the dopamine D1 receptor Cell 111 IssueID2 219–230 Occurrence Handle12408866 Occurrence Handle10.1016/S0092-8674(02)00962-5 Occurrence Handle1:CAS:528:DC%2BD38Xotlahsro%3D
S Balasubramanian JA Teissere DV Raju RA Hall (2004) ArticleTitleHetero-oligomerization between GABAA and GABAB receptors regulates GABAB receptor trafficking J Biol Chem 279 IssueID18 18840–18850 Occurrence Handle14966130 Occurrence Handle10.1074/jbc.M313470200 Occurrence Handle1:CAS:528:DC%2BD2cXjsVKmsbk%3D
E Joo Choi MP Hong Y Kyoo Shin et al. (2003) ArticleTitleATP modulation of sodium currents in rat dorsal root ganglion neurons Brain Res 968 IssueID1 15–25 Occurrence Handle12644260 Occurrence Handle10.1016/S0006-8993(02)04218-X Occurrence Handle1:CAS:528:DC%2BD3sXitV2htb8%3D
Y El-Sherif A Wieraszko P Banerjee NJ Penington (2001) ArticleTitleATP modulates Na+ channel gating and induces a non-selective cation current in a neuronal hippocampal cell line Brain Res 904 IssueID2 307–317 Occurrence Handle11406129 Occurrence Handle10.1016/S0006-8993(01)02487-8 Occurrence Handle1:CAS:528:DC%2BD3MXktlWmsrg%3D
K Dunlap GD Fischbach (1978) ArticleTitleNeurotransmitters decrease the calcium component of sensory neurone action potentials Nature 276 IssueID5690 837–839 Occurrence Handle31570 Occurrence Handle10.1038/276837a0 Occurrence Handle1:CAS:528:DyaE1MXktFChsrw%3D
K Dunlap GD Fischbach (1981) ArticleTitleNeurotransmitters decrease the calcium conductance activated by depolarization of embryonic chick sensory neurones J Physiol 317 519–535 Occurrence Handle6118434 Occurrence Handle1:CAS:528:DyaL3MXlt1KgsL8%3D
M Diverse-Pierluissi K Dunlap EW Westhead (1991) ArticleTitleMultiple actions of extracellular ATP on calcium currents in cultured bovine chromaffin cells Proc Natl Acad Sci USA 88 IssueID4 1261–1265 Occurrence Handle1847515 Occurrence Handle10.1073/pnas.88.4.1261 Occurrence Handle1:CAS:528:DyaK3MXhsVSgur8%3D
L Gandia AG Garcia M Morad (1993) ArticleTitleATP modulation of calcium channels in chromaffin cells J Physiol 470 55–72 Occurrence Handle8308743 Occurrence Handle1:CAS:528:DyaK2cXivVWksQ%3D%3D
KP Currie AP Fox (1996) ArticleTitleATP serves as a negative feedback inhibitor of voltage-gated Ca2+ channel currents in cultured bovine adrenal chromaffin cells Neuron 16 IssueID5 1027–1036 Occurrence Handle8630241 Occurrence Handle10.1016/S0896-6273(00)80126-9 Occurrence Handle1:CAS:528:DyaK28XjsVKgsLs%3D
AD Powell AG Teschemacher EP Seward (2000) ArticleTitleP2Y purinoceptors inhibit exocytosis in adrenal chromaffin cells via modulation of voltage-operated calcium channels J Neurosci 20 IssueID2 606–616 Occurrence Handle10632590 Occurrence Handle1:CAS:528:DC%2BD3cXhtF2hsbw%3D
W Lim SJ Kim HD Yan J Kim (1997) ArticleTitleCa2+-channel-dependent and -independent inhibition of exocytosis by extracellular ATP in voltage-clamped rat adrenal chromaffin cells Pflugers Arch 435 IssueID1 34–42 Occurrence Handle9359901 Occurrence Handle10.1007/s004240050481 Occurrence Handle1:CAS:528:DyaK2sXnt1Grt7Y%3D
KS Elmslie (1992) ArticleTitleCalcium current modulation in frog sympathetic neurones: Multiple neurotransmitters and G proteins J Physiol 451 229–246 Occurrence Handle1357163 Occurrence Handle1:CAS:528:DyaK38XisFals7c%3D
AK Filippov DA Brown (1996) ArticleTitleActivation of nucleotide receptors inhibits high-threshold calcium currents in NG108-15 neuronal hybrid cells Eur J Neurosci 8 IssueID6 1149–1155 Occurrence Handle8752584 Occurrence Handle10.1111/j.1460-9568.1996.tb01282.x Occurrence Handle1:STN:280:DyaK28zisFGmtA%3D%3D
N Vartian S Boehm (2001) ArticleTitleP2Y receptor-mediated inhibition of voltage-activated Ca(2+) currents in PC12 cells Eur J Neurosci 13 IssueID5 899–908 Occurrence Handle11264662 Occurrence Handle10.1046/j.1460-9568.2001.01461.x Occurrence Handle1:STN:280:DC%2BD3M7ms1amug%3D%3D
MB Kulick I Kugelgen Particlevon (2002) ArticleTitleP2Y-receptors mediating an inhibition of the evoked entry of calcium through N-type calcium channels at neuronal processes J Pharmacol Exp Ther 303 IssueID2 520–526 Occurrence Handle12388631 Occurrence Handle10.1124/jpet.102.037960 Occurrence Handle1:CAS:528:DC%2BD38XotlCgurg%3D
H Kubista SG Lechner AM Wolf S Boehm (2003) ArticleTitleAttenuation of the P2Y receptor-mediated control of neuronal Ca2+ channels in PC12 cells by antithrombotic drugs Br J Pharmacol 138 IssueID2 343–350 Occurrence Handle12540525 Occurrence Handle10.1038/sj.bjp.0705037 Occurrence Handle1:CAS:528:DC%2BD3sXhtlSisbY%3D
M Abe T Endoh T Suzuki (2003) ArticleTitleExtracellular ATP-induced calcium channel inhibition mediated by P1/P2Y purinoceptors in hamster submandibular ganglion neurons Br J Pharmacol 138 IssueID8 1535–1543 Occurrence Handle12721109 Occurrence Handle10.1038/sj.bjp.0705174 Occurrence Handle1:CAS:528:DC%2BD3sXktlWisLw%3D
Z Gerevich SJ Borvendeg W Schroder et al. (2004) ArticleTitleInhibition of N-type voltage-activated calcium channels in rat dorsal root ganglion neurons by P2Y receptors is a possible mechanism of ADP-induced analgesia J Neurosci 24 IssueID4 797–807 Occurrence Handle14749424 Occurrence Handle10.1523/JNEUROSCI.4019-03.2004 Occurrence Handle1:CAS:528:DC%2BD2cXhsFOisbg%3D
Lechner SG, Dorostkar MM, Mayer M et al. Autoinhibition of transmitter release from PC12 cells and sympathetic neurons through a P2Y12 receptor-mediated inhibition of voltage-gated Ca2+ channels. Eur J Neurosci (in press).
F Scamps G Vassort (1994) ArticleTitlePharmacological profile of the ATP-mediated increase in L-type calcium current amplitude and activation of a non-specific cationic current in rat ventricular cells Br J Pharmacol 113 IssueID3 982–986 Occurrence Handle7858894 Occurrence Handle1:CAS:528:DyaK2cXmslOmt7o%3D
S Dave DJ Mogul (1996) ArticleTitleATP receptor activation potentiates a voltage-dependent Ca channel in hippocampal neurons Brain Res 715 IssueID1–2 208–216 Occurrence Handle8739640 Occurrence Handle10.1016/0006-8993(95)01588-4 Occurrence Handle1:CAS:528:DyaK28Xisl2lsbo%3D
AK Filippov AA Selyanko J Robbins DA Brown (1994) ArticleTitleActivation of nucleotide receptors inhibits M-type K current [IK(M)] in neuroblastoma × glioma hybrid cells Pflugers Arch 429 IssueID2 223–230 Occurrence Handle7892108 Occurrence Handle10.1007/BF00374316 Occurrence Handle1:CAS:528:DyaK2MXjslanur0%3D
AK Filippov TE Webb EA Barnard DA Brown (1998) ArticleTitleP2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents J Neurosci 18 IssueID14 5170–5179 Occurrence Handle9651200 Occurrence Handle1:CAS:528:DyaK1cXksFarsb0%3D
AK Filippov TE Webb EA Barnard DA Brown (1999) ArticleTitleDual coupling of heterologously-expressed rat P2Y6 nucleotide receptors to N-type Ca2+ and M-type K+ currents in rat sympathetic neurones Br J Pharmacol 126 IssueID4 1009–1017 Occurrence Handle10193782 Occurrence Handle10.1038/sj.bjp.0702356 Occurrence Handle1:CAS:528:DyaK1MXhvVymsbw%3D
AK Filippov DA Brown EA Barnard (2000) ArticleTitleThe P2Y(1) receptor closes the N-type Ca(2+) channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists Br J Pharmacol 129 IssueID6 1063–1066 Occurrence Handle10725253 Occurrence Handle10.1038/sj.bjp.0703185 Occurrence Handle1:CAS:528:DC%2BD3cXit1egsr4%3D
DA Brown AK Filippov EA Barnard (2000) ArticleTitleInhibition of potassium and calcium currents in neurones by molecularly-defined P2Y receptors J Auton Nerv Syst 81 IssueID1–3 31–36 Occurrence Handle10869697 Occurrence Handle10.1016/S0165-1838(00)00150-8 Occurrence Handle1:CAS:528:DC%2BD3cXktlKqtrY%3D
AK Filippov J Simon EA Barnard DA Brown (2003) ArticleTitleCoupling of the nucleotide P2Y4 receptor to neuronal ion channels Br J Pharmacol 138 IssueID2 400–406 Occurrence Handle12540532 Occurrence Handle10.1038/sj.bjp.0705043 Occurrence Handle1:CAS:528:DC%2BD3sXhtlSitrs%3D
PR Adams DA Brown A Constanti (1982) ArticleTitlePharmacological inhibition of the M-current J Physiol 332 223–262 Occurrence Handle6760380 Occurrence Handle1:CAS:528:DyaL38XlsFOjt7k%3D
T Akasu K Hirai K Koketsu (1983) ArticleTitleModulatory actions of ATP on membrane potentials of bullfrog sympathetic ganglion cells Brain Res 258 IssueID2 313–317 Occurrence Handle6600643 Occurrence Handle10.1016/0006-8993(83)91157-5 Occurrence Handle1:CAS:528:DyaL3sXhtVWktr4%3D
HS Lopez PR Adams (1989) ArticleTitleA G protein mediates the inhibition of the voltage-dependent potassium M current by muscarine, LHRH, substance P and UTP in bullfrog sympathetic neurons Eur J Neurosci 1 IssueID5 529–542 Occurrence Handle12106139 Occurrence Handle10.1111/j.1460-9568.1989.tb00360.x
T Tokimasa T Akasu (1990) ArticleTitleATP regulates muscarine-sensitive potassium current in dissociated bull-frog primary afferent neurones J Physiol 426 241–264 Occurrence Handle2121960 Occurrence Handle1:STN:280:DyaK3M%2FjvVylsg%3D%3D
S Boehm (1998) ArticleTitleSelective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors Br J Pharmacol 124 IssueID6 1261–1269 Occurrence Handle9720799 Occurrence Handle10.1038/sj.bjp.0701956 Occurrence Handle1:CAS:528:DyaK1cXltFGlsbc%3D
E Bofill-Cardona N Vartian C Nanoff et al. (2000) ArticleTitleTwo different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides Mol Pharmacol 57 IssueID6 1165–1172 Occurrence Handle10825387 Occurrence Handle1:CAS:528:DC%2BD3cXjvVSjsbk%3D
W Norenberg I Kugelgen Particlevon A Meyer et al. (2000) ArticleTitleM-type K+ currents in rat cultured thoracolumbar sympathetic neurones and their role in uracil nucleotide-evoked noradrenaline release Br J Pharmacol 129 IssueID4 709–723 Occurrence Handle10683196 Occurrence Handle10.1038/sj.bjp.0703096 Occurrence Handle1:CAS:528:DC%2BD3cXhslWqsLo%3D
H Meng M Sakakibara H Nakazawa T Tokimasa (2003) ArticleTitlePyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid can antagonize the purinoceptor-mediated inhibition of M-current in bullfrog sympathetic neurons Neurosci Lett 337 IssueID2 93–96 Occurrence Handle12527396 Occurrence Handle10.1016/s0304-3940(02)01314-9 Occurrence Handle1:CAS:528:DC%2BD3sXivVGgug%3D%3D
Y Ikeuchi T Nishizaki (1995) ArticleTitleATP-evoked potassium currents in rat striatal neurons are mediated by a P2 purinergic receptor Neurosci Lett 190 IssueID2 89–92 Occurrence Handle7644129 Occurrence Handle10.1016/0304-3940(95)11508-T Occurrence Handle1:CAS:528:DyaK2MXls12gu78%3D
Y Ikeuchi T Nishizaki (1995) ArticleTitleThe P2Y purinoceptor-operated potassium channel is possibly regulated by the beta gamma subunits of a pertussis toxin-insensitive G-protein in cultured rat inferior colliculus neurons Biochem Biophys Res Commun 214 IssueID2 589–596 Occurrence Handle7677769 Occurrence Handle10.1006/bbrc.1995.2326 Occurrence Handle1:CAS:528:DyaK2MXotV2ht7w%3D
T Nishizaki Y Ikeuchi (1996) ArticleTitleAdenosine evokes potassium currents by protein kinase C activated via a novel signaling pathway in superior colliculus neurons FEBS Lett 378 IssueID1 1–6 Occurrence Handle8549792 Occurrence Handle10.1016/0014-5793(95)01399-7 Occurrence Handle1:CAS:528:DyaK28Xns1ylug%3D%3D
Y Ikeuchi T Nishizaki (1996) ArticleTitleP2 purinoceptor-operated potassium channel in rat cerebellar neurons Biochem Biophys Res Commun 218 IssueID1 67–71 Occurrence Handle8573178 Occurrence Handle10.1006/bbrc.1996.0013 Occurrence Handle1:CAS:528:DyaK28XltFyitQ%3D%3D
Y Ikeuchi T Nishizaki Y Okada (1996) ArticleTitleRepetitive applications of ATP potentiate potassium current by Ca2+/calmodulin kinase in cultured rat hippocampal neurons Neurosci Lett 203 IssueID2 115–118 Occurrence Handle8834107 Occurrence Handle10.1016/0304-3940(95)12276-1 Occurrence Handle1:CAS:528:DyaK28XlvFSjsg%3D%3D
K Nakazawa K Inoue (1994) ArticleTitleATP reduces voltage-activated K+ current in cultured rat hippocampal neurons Pflugers Arch 429 IssueID1 143–145 Occurrence Handle7708475 Occurrence Handle10.1007/BF02584042 Occurrence Handle1:STN:280:DyaK2M3itlOlsA%3D%3D
P Brown N Dale (2002) ArticleTitleModulation of K(+) currents in Xenopus spinal neurons by p2y receptors: A role for ATP and ADP in motor pattern generation J Physiol 540 IssueIDPt 3 843–850 Occurrence Handle11986373 Occurrence Handle10.1113/jphysiol.2001.013192 Occurrence Handle1:CAS:528:DC%2BD38XjvFelsrg%3D
MD Mark JP Ruppersberg S Herlitze (2000) ArticleTitleRegulation of GIRK channel deactivation by Galpha(q) and Galpha(i/o) pathways Neuropharmacology 39 IssueID12 2360–2373 Occurrence Handle10974320 Occurrence Handle10.1016/S0028-3908(00)00080-0 Occurrence Handle1:CAS:528:DC%2BD3cXmtF2itLg%3D
J Mosbacher R Maier B Fakler et al. (1998) ArticleTitleP2Y receptor subtypes differentially couple to inwardly-rectifying potassium channels FEBS Lett 436 IssueID1 104–110 Occurrence Handle9771902 Occurrence Handle10.1016/S0014-5793(98)01066-7 Occurrence Handle1:CAS:528:DyaK1cXmt1elsrw%3D
K Wirkner L Koles S Thummler et al. (2002) ArticleTitleInteraction between P2Y and NMDA receptors in layer V pyramidal neurons of the rat prefrontal cortex Neuropharmacology 42 IssueID4 476–488 Occurrence Handle11955519 Occurrence Handle10.1016/S0028-3908(01)00199-X Occurrence Handle1:CAS:528:DC%2BD38XivVSisr4%3D
J Luthardt SJ Borvendeg B Sperlagh et al. (2003) ArticleTitleP2Y(1) receptor activation inhibits NMDA receptor-channels in layer V pyramidal neurons of the rat prefrontal and parietal cortex Neurochem Int 42 IssueID2 161–172 Occurrence Handle12421596 Occurrence Handle10.1016/S0197-0186(02)00069-4 Occurrence Handle1:CAS:528:DC%2BD38Xotlymurg%3D
S Ortinau B Laube H Zimmermann (2003) ArticleTitleATP inhibits NMDA receptors after heterologous expression and in cultured hippocampal neurons and attenuates NMDA-mediated neurotoxicity J Neurosci 23 IssueID12 4996–5003 Occurrence Handle12832522 Occurrence Handle1:CAS:528:DC%2BD3sXltlentrs%3D
M Tominaga M Wada M Masu (2001) ArticleTitlePotentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia Proc Natl Acad Sci USA 98 IssueID12 6951–6956 Occurrence Handle11371611 Occurrence Handle10.1073/pnas.111025298 Occurrence Handle1:CAS:528:DC%2BD3MXksVOrsLg%3D
T Moriyama T Iida K Kobayashi et al. (2003) ArticleTitlePossible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity J Neurosci 23 IssueID14 6058–6062 Occurrence Handle12853424 Occurrence Handle1:CAS:528:DC%2BD3sXls1WqsLo%3D
C Vial AB Tobin RJ Evans (2004) ArticleTitleG-protein-coupled receptor regulation of P2X(1) receptors does not involve direct channel phosphorylation Biochem J 382 IssueIDPt 1 101–110 Occurrence Handle15144237 Occurrence Handle1:CAS:528:DC%2BD2cXmsVChsbs%3D
CF Stevens (2004) ArticleTitlePresynaptic function Curr Opin Neurobiol 14 IssueID3 341–345 Occurrence Handle15194114 Occurrence Handle10.1016/j.conb.2004.04.004 Occurrence Handle1:CAS:528:DC%2BD2cXkvV2nsrg%3D
A Meir S Ginsburg A Butkevich et al. (1999) ArticleTitleIon channels in presynaptic nerve terminals and control of transmitter release Physiol Rev 79 IssueID3 1019–1088 Occurrence Handle10390521 Occurrence Handle1:CAS:528:DyaK1MXksFemtrk%3D
PD Dodson ID Forsythe (2004) ArticleTitlePresynaptic K+ channels: Electrifying regulators of synaptic terminal excitability Trends Neurosci 27 IssueID4 210–217 Occurrence Handle15046880 Occurrence Handle10.1016/j.tins.2004.02.012 Occurrence Handle1:CAS:528:DC%2BD2cXisVGiurg%3D
C Luscher LY Jan M Stoffel et al. (1997) ArticleTitleG protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons Neuron 19 IssueID3 687–695 Occurrence Handle9331358 Occurrence Handle10.1016/S0896-6273(00)80381-5 Occurrence Handle1:CAS:528:DyaK2sXmvVKlsLc%3D
D Kristufek G Koth A Motejlek et al. (1999) ArticleTitleModulation of spontaneous and stimulation-evoked transmitter release from rat sympathetic neurons by the cognition enhancer linopirdine: Insights into its mechanisms of action J Neurochem 72 IssueID5 2083–2091 Occurrence Handle10217288 Occurrence Handle10.1046/j.1471-4159.1999.0722083.x Occurrence Handle1:CAS:528:DyaK1MXis12ltL4%3D
SG Lechner M Mayer S Boehm (2003) ArticleTitleActivation of M1 muscarinic receptors triggers transmitter release from rat sympathetic neurons through an inhibition of M-type K+ channels J Physiol 553 IssueIDPt 3 789–802 Occurrence Handle14555721 Occurrence Handle10.1113/jphysiol.2003.052449 Occurrence Handle1:CAS:528:DC%2BD2cXosVSkug%3D%3D
M Martire P Castaldo M D’Amico et al. (2004) ArticleTitleM channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals J Neurosci 24 IssueID3 592–597 Occurrence Handle14736843 Occurrence Handle10.1523/JNEUROSCI.3143-03.2004 Occurrence Handle1:CAS:528:DC%2BD2cXht1Cjtb0%3D
HS Engelman AB MacDermott (2004) ArticleTitlePresynaptic ionotropic receptors and control of transmitter release Nat Rev Neurosci 5 IssueID2 135–145 Occurrence Handle14735116 Occurrence Handle10.1038/nrn1297 Occurrence Handle1:CAS:528:DC%2BD2cXks1CrsQ%3D%3D
RA Cunha JA Ribeiro (2000) ArticleTitleATP as a presynaptic modulator Life Sci 68 IssueID2 119–137 Occurrence Handle11191632 Occurrence Handle10.1016/S0024-3205(00)00923-1 Occurrence Handle1:CAS:528:DC%2BD3cXoslWmsrY%3D
JM Zhang HK Wang CQ Ye et al. (2003) ArticleTitleATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression Neuron 40 IssueID5 971–982 Occurrence Handle14659095 Occurrence Handle10.1016/S0896-6273(03)00717-7 Occurrence Handle1:CAS:528:DC%2BD3sXhtVWgtLbN
GD Price SJ Robertson FA Edwards (2003) ArticleTitleLong-term potentiation of glutamatergic synaptic transmission induced by activation of presynaptic P2Y receptors in the rat medial habenula nucleus Eur J Neurosci 17 IssueID4 844–850 Occurrence Handle12603274 Occurrence Handle10.1046/j.1460-9568.2003.02501.x
L Stjarne P Astrand (1985) ArticleTitleRelative pre- and postjunctional roles of noradrenaline and adenosine 5′-triphosphate as neurotransmitters of the sympathetic nerves of guinea-pig and mouse vas deferens Neuroscience 14 IssueID3 929–946 Occurrence Handle2859555 Occurrence Handle10.1016/0306-4522(85)90155-1 Occurrence Handle1:STN:280:DyaL2M7ovVKkug%3D%3D
I Kugelgen Particlevon K Kurz K Starke (1993) ArticleTitleAxon terminal P2-purinoceptors in feedback control of sympathetic transmitter release Neuroscience 56 IssueID2 263–267 Occurrence Handle10.1016/0306-4522(93)90330-I
S Boehm (2003) ArticleTitleSignaling via nucleotide receptors in the sympathetic nervous system Drug News Perspect 16 IssueID3 141–148 Occurrence Handle12819812 Occurrence Handle10.1358/dnp.2003.16.3.876887 Occurrence Handle1:CAS:528:DC%2BD3sXmtFWit7c%3D
S Boehm H Kubista (2002) ArticleTitleFine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors Pharmacol Rev 54 IssueID1 43–99 Occurrence Handle11870260 Occurrence Handle10.1124/pr.54.1.43 Occurrence Handle1:CAS:528:DC%2BD38XislOgtb4%3D
G Queiroz C Talaia J Goncalves (2003) ArticleTitleATP modulates noradrenaline release by activation of inhibitory P2Y receptors and facilitatory P2X receptors in the rat vas deferens J Pharmacol Exp Ther 307 IssueID2 809–815 Occurrence Handle12966150 Occurrence Handle10.1124/jpet.103.054809 Occurrence Handle1:CAS:528:DC%2BD3sXos1Shsb4%3D
SJ Borvendeg Z Gerevich C Gillen P Illes (2003) ArticleTitleP2Y receptor-mediated inhibition of voltage-dependent Ca2+ channels in rat dorsal root ganglion neurons Synapse 47 IssueID2 159–161 Occurrence Handle12454954 Occurrence Handle10.1002/syn.10156 Occurrence Handle1:CAS:528:DC%2BD3sXlt1Chtg%3D%3D
F Nakamura SM Strittmatter (1996) ArticleTitleP2Y1 purinergic receptors in sensory neurons: Contribution to touch-induced impulse generation Proc Natl Acad Sci USA 93 IssueID19 10465–10470 Occurrence Handle8816824 Occurrence Handle10.1073/pnas.93.19.10465 Occurrence Handle1:CAS:528:DyaK28XlslGrsrY%3D
HZ Hu N Gao MX Zhu et al. (2003) ArticleTitleSlow excitatory synaptic transmission mediated by P2Y1 receptors in the guinea-pig enteric nervous system J Physiol 550 IssueIDPt 2 493–504 Occurrence Handle12807993 Occurrence Handle10.1113/jphysiol.2003.041731 Occurrence Handle1:CAS:528:DC%2BD3sXmsFGktbc%3D
GP Connolly PJ Harrison TW Stone (1993) ArticleTitleAction of purine and pyrimidine nucleotides on the rat superior cervical ganglion Br J Pharmacol 110 IssueID4 1297–1304 Occurrence Handle8306068 Occurrence Handle1:CAS:528:DyaK2cXmvFCmtg%3D%3D
S Boehm S Huck P Illes (1995) ArticleTitleUTP- and ATP-triggered transmitter release from rat sympathetic neurones via separate receptors Br J Pharmacol 116 IssueID5 2341–2343 Occurrence Handle8581264 Occurrence Handle1:CAS:528:DyaK2MXptlersbk%3D
N Vartian E Moskvina T Scholze et al. (2001) ArticleTitleUTP evokes noradrenaline release from rat sympathetic neurons by activation of protein kinase C J Neurochem 77 IssueID3 876–885 Occurrence Handle11331416 Occurrence Handle10.1046/j.1471-4159.2001.00290.x Occurrence Handle1:CAS:528:DC%2BD3MXjs1Wlsbk%3D
SJ Robertson SJ Ennion RJ Evans FA Edwards (2001) ArticleTitleSynaptic P2X receptors Curr Opin Neurobiol 11 IssueID3 378–386 Occurrence Handle11399438 Occurrence Handle10.1016/S0959-4388(00)00222-1 Occurrence Handle1:CAS:528:DC%2BD3MXks1Okur0%3D
Acknowledgement
Work in the authors’ laboratory is supported by grants from the Austrian Science Fund, FWF (P14951 and P15797), and from the Virologiefonds of the Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Lechner, S.G., Boehm, S. Regulation of neuronal ion channels via P2Y receptors. Purinergic Signalling 1, 31–41 (2004). https://doi.org/10.1007/s11302-004-4746-3
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s11302-004-4746-3