
Abstract
Leptospermum scoparium is the basis of a flourishing honey industry in Aotearoa New Zealand (NZ) and Australia. The genetic structure of L. scoparium across its range in NZ and Australia was previously assessed using pooled, whole genome sequencing; however, only one sampling site in Tasmania was included. Here, we used a single nucleotide polymorphism (SNP) array for genotyping samples of L. scoparium collected in natural stands around Tasmania and NZ, to determine the genetic relationship between L. scoparium individuals from the two regions. In total, 2069 high quality, polymorphic SNP markers were applied across the sample set of 504 individuals, revealing that Tasmanian L. scoparium are genetically distinct from NZ mānuka, confirming the observation from the pooled whole genome sequencing project. FST and discriminant analysis of principal components confirmed that the Tasmanian populations are well differentiated genetically from NZ populations, suggesting that they should be recognised as a separate, endemic Australian species. Within NZ, eight geographic groups are distinguished with genotypic variation exhibiting north to south landscape scale patterns with regional genetic clusters. We found support for isolation by distance, and this was reflected in the range of pairwise FST values estimated between NZ genetic clusters (0.056 to 0.356); however, each geographic genetic group exhibits geneflow and is only weakly differentiated from neighbouring clusters as evidenced by low population differentiation (low pairwise FST). These data provide little support for taxonomic revision and subdividing L. scoparium into segregate species within NZ.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leptospermum scoparium J.R.Forst. & G.Forst. is a woody tree species, native to Aotearoa New Zealand (NZ) and forms the basis of a flourishing honey industry, attracting premium prices due to its unique non-peroxide antimicrobial properties (Ministry for Primary Industries 2018). The species is known as mānuka by Māori who have many traditional uses for it and consider it a taonga (culturally significant/treasure). Chemical, morphological, and genetic studies have indicated geographic variation of mānuka across its range in NZ and genetic differentiation between NZ and Australia (Ronghua et al. 1984; Perry et al. 1997; Williams et al. 2014; Koot et al. 2022). Pooled whole genome re-sequencing of 76 L. scoparium and outgroup populations from NZ and Australia (Koot et al. 2022) previously determined the genetic structure and relatedness of L. scoparium across NZ, as well as between populations in NZ and Australia. Population genomic investigation of this dataset, consisting of ~ 2.5 million single nucleotide polymorphisms (SNPs), suggested that there are five geographically and genetically distinct mānuka clusters within NZ, with evidence of gene flow occurring between these clusters. Mānuka populations in NZ are genetically distinct from populations in Australia, with coalescent modelling suggesting that these two clades diverged ~ 9–12 million years ago. However, the research by Koot et al. (2022) included only a single sampling site from Tasmania, a region where L. scoparium grows naturally and abundantly and contributes to local honey production. Here we addressed this sampling gap by increasing the representation of L. scoparium samples from across Tasmania, including from the eastern, northern, and western coasts of Tasmania. We also increased the representation of NZ populations by sampling over a wider geographic area, filling in sampling gaps of the Koot et al. 2022 study.
The study published by Koot and colleagues used a method based on pooling DNA samples from each site (i.e., 30 L. scoparium trees sampled and pooled for each of the 76 sites) and then performing whole genome sequencing at high depth (> 160 × coverage of the full genome). The pooled sequencing method is well recognised as capable of estimating DNA variant frequencies, genetic structure, and diversity within and among populations, and it can be used to calculate genetic differentiation using FST metrics (Futschik and Schlötterer 2010; Schlötterer et al. 2014). Genotyping using markers screened on individual samples (instead of pools) can help confirm the results obtained by the pooled whole genome sequencing method. Hundreds of DNA markers distributed across the genome are typically applied for such population genetics analysis. Here, we implemented a new method based on a SNP array capable of screening nine thousand DNA markers simultaneously. SNP arrays are a widely used technology for genotyping DNA of many organisms, including animals and plants for selective breeding or pedigree analysis, as well as elucidating phylogeographic patterns and taxonomic structure (Montanari et al. 2022).
Leptospermum scoparium has traditionally been interpreted as a naturally variable species in NZ. While taxa L. scoparium var. scoparium and L. scoparium var. incanum Cockayne have been accepted (Cockayne 1917; Allan 1961), Thompson (1983) recognised only L. scoparium. More recently, the species L. repo has been formally recognised (de Lange and Schmid 2021). Remarkably, and despite only three (Buys et al. 2019) and five (Koot et al. 2022) genetic groups being distinguished, there remain fourteen other morphotypes provisionally recognised from NZ (Li et al. 2022). These morphotypes are claimed to represent potential unnamed species based on their growth habit, leaf shape, flower size and colour, and capsule shape, size and colour (Li et al. 2022). However, they are not scientifically named and described (i.e., with diagnostic characters and known distributions) making it difficult to independently test them with genetic data. Our approach in this study is to determine whether genetically discrete metapopulation lineages (sensu de Queiroz 2007) of NZ Leptospermum can be retrieved.
This research describes the genetic variability of wild populations of L. scoparium across their natural range. Collection of L. scoparium samples from Tasmania was genotyped with a SNP array to confirm that L. scoparium populations from NZ and Australia are differentiated genetically. To address variation among NZ populations, our approach was to utilise these SNP data to recover any metapopulation genetic lineages that require further systematics research. Furthermore, additional sampling of NZ populations provided improved phylogeographic resolution compared to previous studies.
Material and methods
Sampling and DNA extraction
In total, 720 samples (Supplementary Table 1) were collected from 24 locations across Tasmania, Australia (Supplementary Fig. 1). The sampling was done in 96-well format, with the plates placed in a Styrofoam box with silica beads at the bottom of the wells to allow desiccation of the leaves on site. In total, eight 96-well plates were sampled. Between 10 and 15 leaves were collected in each well. Young expanding leaves at the tip of actively growing shoots were picked and up to 30 individuals per site were identified and sampled, ensuring as much distance as possible between sampled trees to avoid collecting siblings. Plates were then sealed and shipped to Intertek, Adelaide, Australia, for DNA extraction using their automated plant DNA protocol. Intertek reported DNA concentrations of approximately 10 ng/µL for the eight plates, with 1 µg of DNA per sample. DNA samples were dried and shipped to Labogena, Jouy-en-Josas, France, for SNP array genotyping.
A total of 264 DNA samples from the Koot et al. (2022) study, representative of three NZ provenances (Northland, East Cape and south-western South Island) were available for genotyping (Supplementary Table 2). The DNA samples that had already been extracted for the whole genome sequencing were aliquoted, dried, and sent for genotyping at Labogena. Informed consent was obtained from the Māori governance group listed in Koot et al. (2022) for re-using these DNA samples for the purpose of this study. A further set of 192 samples was obtained from 22 locations across NZ, including one location from Chatham Islands. Samples were collected in zip-lock bags containing silica gel. DNA was manually extracted using the Macherey–Nagel Plant II extraction kit, using the PL2 extraction buffer. DNA was quantified with the Quant-iT™ PicoGreen dsDNA assay (Invitrogen), which revealed an average concentration of 18 ng/µL and 0.7 µg per sample.
SNP array genotyping
The array technology was the Applied Biosystems® Axiom™ system (Thermo-Fisher Scientific) and the array design and evaluation are described by Montanari et al. (2022). The Axiom array genotyping was performed by Labogena, using standard protocols. The array contained 9002 SNPs for mānuka, as well as SNPs for other species (the fish trevally and snapper, and the fruits raspberry and blackberry). Bioinformatics analyses of the DNA sequences flanking the mānuka SNPs were carried out at the array design stage to verify that mānuka DNA would not hybridise with the markers targeting other species. All mānuka samples screened were mixed with snapper DNA for multiplexing as described and evaluated by Montanari et al. (2022). No replicate samples were included between experimental units (96-well plates). After genotyping, the SNP array data were analysed using the Axiom Analysis Suite v5.1.1 software applying a dish QC threshold of 0.82 and a QC Call Rate > 95. Visual inspection of SNP cluster plots was performed for SNPs classified as ‘PolyHighResolution’ (PHR) and ‘CallRateBelowThreshold’ (CRBT) in the Axiom Analysis Suite software, and SNP calls were manually curated. During this manual curation, SNPs for which genotypic clusters were not clearly defined were removed. Specifically, SNPs that had two distinct groups of samples (corresponding to different sampling batches) assigned to the same genotypic clusters were discarded.
Genetic diversity analysis
Population structure was explored using k-means clustering (find.clusters) and discriminant analysis of principal components (DAPC) functions from the R package adegenet v2.1.1 (Jombart et al. 2010; Jombart and Ahmed 2011). The k-means clustering analysis was carried out using the following parameters: max.n = 20, n.pca = 500, scale = FALSE, choose.n.clust = FALSE, criterion = “min”. An initial DAPC analysis was then performed, applying: grp = grp$grp, n.pca = 100, n.da = 100, scale = FALSE, var.contrib = FALSE. Subsequently, an optimum number of principal components (PCs) was determined from this initial run using the adegenet optim.a.score function. A final DAPC run was then carried out, applying both optimized K and PC values. Additionally, population structure was explored using the R packages LEA (Frichot and François 2015) and tess3r (Caye et al. 2016) applying the snmf and tess3 functions, respectively. The following parameters were used for the snmf analysis: K = 1:15, entropy = T, ploidy = 2, project = “new”, repetitions = 10, tolerance = 0.00001. And the following parameters were used for the tess3() analysis: K = 1:15, method = “projected.ls”, ploidy = 2.
After establishing the number of clusters in the dataset, the R package dartR v2.0.3 (Mijangos et al. 2022) was used to estimate summary statistics for each cluster applying the gl.basic.stats function, and outputting estimates of mean observed heterozygosities (Ho), mean gene diversities within population (Hs) and inbreeding coefficients (Fis). The R package StAMPP v1.6.3 (Pembleton et al. 2013) was used to estimate individual pairwise Nei’s genetic distances (stamppNeisD) (Nei 1972), and population pairwise fixation indices (FST) (stamppFst) (Weir and Cockerham 1984), using the clusters identified across analyses as the populations for comparison. An unrooted Neighbor-net network and Isolation by Distance (IBD) analyses were implemented in SplitsTree4 v4.14.8 (Huson and Bryant 2005) and ade4 v1.7–15 (Dray and Dufour 2007), respectively, both using the Nei’s genetic distances estimated in StAMPP. TreeMix v1.13 (Pickrell and Pritchard 2012) was used to investigate migration and historical relationships between populations. Ten migration events were explored for five iterations, and the trees were rooted by Tasmanian samples. The R package OptM v0.1.3 (Fitak 2021) was then used to estimate the optimal number of migration edges that would best explain the dataset, applying the evanno method and a threshold of 0.05.
The relationship between NZ clusters and the environment was explored using a permutational multivariate analysis of variance (PERMANOVA). Data for seven environmental variables were extracted from the New Zealand Environmental Data Stack (McCarthy et al. 2021): annual precipitation, annual solar radiation, October vapour pressure deficit, mean annual temperature, mean temperature of the coldest quarter, potential evapotranspiration ratio and soil particle size. The contribution of environmental variables was investigated using principal components analysis (PCA), applying the R stats v3.6.2 prcomp function. The function adornis2 from the R package vegan v2.6–4 (Dixon 2003) and the package pairwiseAdonis v0.4 (Martinez Arbizu 2017) were used to run permutational multivariate analysis of variance analyses (PERMOVAs), testing for significant differences between clusters. Each analysis was run for 100,000 permutations and Bonferroni correction methods were applied.
Data sovereignty statement
Mānuka is considered taonga (treasured entity) by Māori. The notion of taonga describes physical entities such as plants and wildlife, but also cultural works, and intangible things such as language, haka, identity and culture. As a taonga, Māori have traditional responsibilities of guardianship (kaitiakitanga) over mānuka. Informed consent was granted by Māori landowners to re-use the reference genome of L. scoparium ‘Crimson Glory’ (Thrimawithana et al. 2019), as well as the pool-sequencing data and DNA samples of Koot et al. (2022), for the purpose of analysing the genetic difference between NZ and Australia L. scoparium. Due to the sensitivity of the indigenous intellectual property of these samples, exact sampling sites and the identities of those who contributed material are excluded, and only regions of origin will be used to describe sampling locations.
Results
SNP array analysis
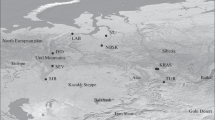
Of the total 9002 mānuka SNPs on the array, 3003 and 1861 were classified as PHR and CRBT using the Axiom Analysis Suite software, respectively. Following manual curation, 2069 SNPs were kept for further analysis. Of the full dataset of 1182 L. scoparium samples, 418 samples from NZ (92.5% of 456 in total, including 43 L. repo), and 86 samples from Tasmania (11.9% of 720 in total; Supplementary Table 3) — totalling 504 samples, produced successful genotyping results (Fig. 1; Table 1). The 86 Tasmanian samples that were successfully genotyped represented a distribution of locations around Tasmania, and the data were therefore used for further analysis of genetic diversity.

Map of sampling locations in Aotearoa New Zealand and Tasmania (Australia). Sampling sites are named according to their region of origin and points are purposefully enlarged to address the sensitivity of this indigenous intellectual property
Genetic diversity analysis
The k-means clustering analysis determined there to be ten genetically and geographically distinct clusters within the dataset (Fig. 2A). Of the 20 K values explored by the k-means clustering analysis, K = 10 had the lowest Bayesian information criterion (BIC) value (BIC = 3273), followed by K = 9 (BIC = 3275.3) and K = 8 (BIC = 3275.6) (Supplementary Table 4). The DAPC analysis using this optimal K value supported the segregation of the 504 samples into ten clusters (Fig. 2B). Linear dimensions (LD) one, two and three (LD1, LD2, and LD3) of the DAPC (K = 10) explained 65.8%, 13.7% and 9.1% of the data variation, respectively (Supplementary Table 5). Along LD1, clear separation between NZ and Tasmanian samples is visible, as well as the separation of Tasmanian samples into two clusters. Along LD2 and LD3, NZ samples form clusters that closely match their geographical distribution, with LD2 separating samples north–south, and LD3 separating samples east–west. The two Tasmanian clusters represent samples from northern and western Tasmania (NWT) and southern and eastern Tasmania (SET), while the eight NZ clusters comprise samples from northern Northland (NNI1 and NNI2), central and southern North Island (CSNI), East Cape North Island (ECNI), northern South Island (NSI), north-eastern South Island (NESI), south-western South Island (SWSI) and a cluster of L. repo samples from central North Island (Table 1). There were four notable instances where a sample did not group with the genetic cluster it was predicted to, based on geographic location and genetic clusters established by Koot et al. 2022. This includes two samples from the West Coast 2 population clustering with CSNI (instead of SWSI); a sample from Canterbury 9 that clustered with NESI (instead of SWSI), a sample from Southland 2 that clustered with ECNI (instead of SWSI) and a sample from Wellington 1 that clustered with L. repo (instead of CSNI) (Table 1).

Population structure and admixture of Aotearoa New Zealand (NZ) and Tasmanian Leptospermum scoparium and L. repo. LD: Linear Dimension; SWSI: Southwest South Island (NZ); NNI2: northern North Island subcluster #2 (NZ); NWT: northwest Tasmania (Australia); CSNI: central and southern North Island (NZ); ECNI: East Cape North Island (NZ); SET: southeast Tasmania (Australia); NNI1: northern North Island subcluster #1 (NZ); NESI: northeast South Island (NZ), NSI: northern South Island (NZ); and CI: Chatham Islands (NZ). (A) Bayesian information criterion (BIC) plotted against the number of clusters (K) tested. The optimal number of clusters is indicated by the lowest BIC value (K = 10). (B) Discriminant analysis of principal components (DAPC) of 504 samples of L. scoparium and L. repo. (C) Lea admixture analysis with number of clusters ranging from K = 9 to K = 11
LEA ancestral admixture was explored across three K values (K = 9 to K = 11) (Fig. 2C), based on the k-means clustering result. When K = 9, eight ancestral clusters were identified within the NZ sampling, with all Tasmanian samples forming one distinct cluster. The eight NZ clusters comprise samples from NNI1, NNI2, CSNI, ECNI, NESI, SWSI, the Chatham Islands (CI) and a cluster of L. repo samples. When K = 10, the eight NZ clusters remain, with an additional cluster emerging within the Tasmanian samples, splitting them into NWT and SET. When K = 11, an additional cluster within the South Island samples emerges, representing samples from Canterbury 1 and Tasman in northern South Island (NSI). Admixture between NN1 and NNI2, NNI1 and CSNI, NNI1 and L. repo, CSNI and ECNI, CSNI and L. repo, NESI and CSNI, NESI and L. repo, NESI and CI, and low levels of admixture between SWSI and all other clusters is apparent across all K values — and largely reflects introgression between geographically neighbouring clusters.
Structure within NZ samples was further explored using DAPC and tess3r (Fig. 3). The DAPC of just NZ samples identified seven clusters within the dataset — NNI1, NNI2, CSNI, ECNI, NESI, SWSI and L. repo, with LD1, LD2, and LD3 of the DAPC (K = 7) explaining 43.0%, 30.0% and 11.0% of the data variation, respectively (Fig. 3A). Results for K = 7 to K = 9 of the tess3r analysis were explored, with K = 7 identifying the same seven clusters as the DAPC anlysis (Fig. 3c). K = 8 performed similarly to the K = 9 LEA admixture analysis, with CI samples splitting off into their own cluster. When K = 9, the South Island samples formed a third cluster (NSI), as seen in the LEA K = 11 results. As with the LEA admixture analysis, admixture was apparent between NNI1 and NNI2, NNI1 and CSNI, NNI and L. repo, CSNI and ECNI, CSNI and L. repo, NESI and CSNI, NESI and L. repo, and low levels of admixture between SWSI and all other clusters is apparent across all K values (Fig. 3C). The results of the tess3r analysis were projected across maps of NZ, so that the geographic structuring of the clusters can be more easily visualised (Fig. 3B).

Population structure and admixture of Aotearoa New Zealand (NZ) Leptospermum scoparium and L. repo. LD: Linear Dimension; SWSI: Southwest South Island (NZ); NNI2: northern North Island subcluster #2 (NZ); CSNI: central and southern North Island (NZ); ECNI: East Cape North Island (NZ); NNI1: northern North Island subcluster #1 (NZ); NESI: northeast South Island (NZ), CI: Chatham Islands (NZ). (A) Discriminant analysis of principal components (DAPC) of 422 samples of L. scoparium and L. repo from NZ. (B) Geographical distribution of NZ clusters ranging from K = 7 to K = 9. Note that the Chatham Islands are not visible on this map. (C) Admixture analysis of NZ clusters using the tess3r package ranging from K = 7 to K = 9
Nei’s genetic distances were estimated between individuals and the distance matrix was subsequently used to produce an unrooted SplitsTree Neighbor-net network (Fig. 4). Clear divergence between NZ and Tasmanian samples is visible. NZ samples form clusters matching similarly to those determined by k-means clustering, DAPC and admixture analyses, although clustering is not always as distinct (e.g. SWSI, NESI, and NSI; NNI1 an L.repo; NESI; CSNI) and conflicting signal is evident in the central boxes formed within the network. There are two samples from CSNI that sit away from other CSNI samples but closely with NESI. These two CSNI samples are from the West Coast of the South Island (West Coast 2) but clustered with CSNI samples in the DAPC. The network suggests that NNI2 is derived from NNI1, and similarly ECNI from CSNI. The Nei’s genetic distance matrix was also used to run an IBD analysis across NZ samples, with a significant relationship being identified between genetic and geographic distances (R2 = 0.2165, p-value = 0.001).

Unrooted Neighbor-net network of Leptospermum scoparium and L. repo from Aotearoa New Zealand (NZ) and Tasmania (Australia) calculated using Nei’s genetic distance, coloured by k-means cluster. NWT: northwest Tasmania (Australia); SET: southeast Tasmania (Australia); NESI: northeast South Island (NZ); CSNI: central and southern North Island (NZ); ECNI: East Cape North Island (NZ); NNI1: northern North Island subcluster #1 (NZ); NNI2: northern North Island subcluster #2 (NZ); SWSI: Southwest South Island (NZ); NSI: northern North Island (NZ)
Summary statistics and pairwise FST distances were estimated based on the ten clusters identified by the k-means clustering analysis. As cluster sizes varied greatly, a random subset of individuals from each cluster was selected, so that each cluster subset had the same number of individuals as the smallest cluster (n = 14) for genetic diversity estimation. Observed heterozygosity (HO) ranged from 0.278 (SWSI) to 0.292 (NSI) within the NZ samples, and 0.286 (NWT) to 0.290 (SET) within the Tasmanian samples (Table 2). Within population gene diversity (HS) ranged from 0.185 (SWSI) to 0.203 (NNI1) within the NZ samples, and from 0.181 (NWT) to 0.186 (SET) within the Tasmanian samples. And observed inbreeding coefficients (FIS) ranged from − 0.582 (NSI) to − 0.430 (NNI1) within the NZ sampling, and from − 0.576 (NWT) to − 0.563 (SET) within the Tasmanian samples. Mean pairwise FST distances between NZ clusters was 0.21, and FST between the two Tasmanian populations was 0.14 (Fig. 5). Pairwise FST within NZ samples ranged from 0.062 (NESI and SWSI) to 0.365 (NSI and NNI2). Mean FST distance between NZ and Tasmanian clusters was 0.33. Pairwise FST between NZ and Tasmanian clusters ranged from 0.27 (SET and NNI1) and 0.39 (NWT and ECNI). All pairwise FST estimates were statistically significant (p-values = < 0.001).

A heatmap of pairwise FST values for ten clusters of Leptospermum scoparium and L. repo in Aotearoa New Zealand (NZ) and Tasmania (Australia). SWSI: Southwest South Island (NZ); NESI: northeast South Island (NZ); NSI: northern South Island (NZ), NNI2: northern North Island subcluster #2 (NZ); NWT: northwest Tasmania (Australia); CSNI: central and southern North Island (NZ); NNI1: northern North Island subcluster #1 (NZ); SET: southeast Tasmania (Australia); ECNI: East Cape North Island (NZ). Dark blue coloured squares reflect low FST values, and red coloured squares reflect high FST values
TreeMix was used to explore migration between clusters. Of the 10 migration events explored, OptM determined four to be the optimal number of migration events that best explained the dataset, with a Delta m score of 4.51 and mean f of 0.99 (Fig. 6A; Supplementary Table 6). The four migration edges identified by TreeMix were from NWT to the NNI2 cluster, NNI1 to L. repo, CSNI/ECNI to NNI1 and from CSNI/ECNI to NESI, with no migration edges occurring from NZ back to Tasmania. The Maximum Likelihood (ML) tree produced by TreeMix (m = 4) was rooted by the Tasmanian cluster NWT, with both Tasmanian clusters remaining separate from the NZ clusters (Fig. 6B). The ML tree places the two clusters from Northland (NNI1 and NNI2) as a sister clade to the remaining six clusters from NZ, while SWSI, NSI and NESI form a clade sister to CSNI, ECNI and L. repo.

TreeMix genetic migration analysis of ten clusters of Leptospermum scoparium and L. repo from Aotearoa New Zealand (NZ) and Tasmania (Australia). (A) Graph of Delta m and mean f scores for 10 migration events modelled by TreeMix analysis. (B) Maximum likelihood (ML) phylogenetic tree of L. scoparium and L. repo samples for m = 4. Colour of migration edges (arrows) indicates significance of migration event: yellow = less significant, red = very significant. SWSI: Southwest South Island (NZ); NSI: northern South Island (NZ); NESI: northeast South Island (NZ); CSNI: central and southern North Island (NZ); ECNI: East Cape North Island (NZ); NNI2: northern North Island subcluster #2 (NZ); NNI1: northern North Island subcluster #1 (NZ); SET: southeast Tasmania (Australia); NWT: northwest Tasmania (Australia). C) Heatmap of residual fit from TreeMix ML tree
Environmental effect
Principal component analysis dimensions one (Dim1), two (Dim2) and three (Dim3) explained 52.0%, 22.6% and 11.7% of variance within the environmental dataset, respectively (Fig. 7). Mean annual temperature (25.4%), potential evapotranspiration ratio (23.8%) and mean temperature of the coldest quarter (23.3%) made the greatest contribution towards Dim1, with annual precipitation (0.34%) and soil particle size (0.0003%) making the lowest contributions. Annual precipitation (41.5), October vapour pressure deficit (29.2%) and soil particle size (27.8%) made the greatest contributions towards Dim2, and annual solar radiation (0.05%) and mean annual temperature (0.04%) made the lowest contributions. A PCA of the SNP dataset was also carried out for comparison, with Dim1, Dim2 and Dim3 explaining 13.3%, 7.12% and 2.7% of variance within the dataset, respectively. Results of the PERMOVA revealed there is a significant relationship between clusters and the environmental variables (R2 = 0.278, F = 156.46, p-value = 9.99E-04). When explored further via pairwise comparisons of clusters, all 28 comparisons were found to be significant (p-values = < 0.05; Supplementary Table 7).

PCA of seven environmental variables for Leptospermum scoparium and L. repo in Aotearoa New Zealand. (A) Bar plot of explained variance for first seven principal components of PCA. (B) PCA biplot of environmental variables, with variables coloured by level of contribution — with red indicating a greater level of contribution, and blue indicating less contribution
Discussion
Strong genetic differentiation between Aotearoa New Zealand and Tasmanian L. scoparium populations
Our population genetic analysis of L. scoparium using 2069 SNPs indicated a strong genetic differentiation between NZ and Australian populations. The results from the DAPC and FST analysis reported in this study, which included samples from multiple locations in Tasmania, are similar to the results obtained by Koot et al. (2022) that used pooled whole genome re-sequencing. However, the results presented here represent a wider distribution of L. scoparium samples from Tasmania than in the Koot et al. (2022) study, expanding from one site in Tasmania, to seven sites from the west, northwest, east and southeast of Tasmania. In both Koot et al. (2022) and this new study, the DAPC and FST analyses clearly discriminated L. scoparium populations from NZ and Australian origins. The FST values measured by both approaches indicate very high genetic differentiation. FST values greater than 0.25 are widely accepted as being indicative of very different genetic stocks (Hartl et al. 1997; Frankham et al. 2002). Taken together, these findings confirm that the Tasmanian populations are genetically distinct from NZ populations, which provides evidence that they should be recognised as an endemic Australian species separate from L. scoparium, and subsequently L. scoparium be treated as endemic to NZ where the type specimen is from. The taxonomic consequences of variation within the Tasmanian samples require further investigation with two subclusters being distinguished (Fig. 2, 4).
Taxonomic considerations of Aotearoa New Zealand populations
Compared with earlier studies (Buys et al. 2019; Koot et al. 2022), our increased sampling across a wide geographic area provides new insights and contributes to two taxonomic outcomes for NZ mānuka. First, L. scoparium is a NZ endemic metapopulation lineage and, second, there is no support for taxonomic recognition among NZ populations for either L. repo or any of the other morphological segregates provisionally recognised (de Lange and Schmid 2021; Li et al. (2022);. Recognition of these morphological segregates is perplexing as none are recovered as groups by Buys et al. (2019), Koot et al. (2022) or in this study. It would be expected that if any taxonomic group exist, they would be recovered in this study of genome-wide SNPs. While the ancestral admixture analyses (Fig. 2C, 3C) indicated groupings among NZ populations of L. scoparium and L. repo, the DAPC (Fig. 2B, 3A), Neighbor-net network (Fig. 4), pairwise FST (Fig. 5) and TreeMix (Fig. 6) analyses showed detailed aspects of close relationships among these groups. A case in point is the low genetic differentiation (FST values) of L. repo from adjacent northern North Island (FST = 0.1–0.18) and central and southern North Island populations (FST = 0.2) (Fig. 5). Moreover, in the Neighbor-net analysis the reticulate central area of the network and long edges connecting L. repo to northern North Island or central and southern North Island populations attests to ambiguity regarding its distinctiveness and clarity of relationships (Fig. 4), as does the overlap of L. repo with northern North Island populations in the DAPC (Fig. 2A), and migration events with NNI1 demonstrated by the TreeMix analysis (Fig. 6). Similar can be said for clusters identified from the South Island (e.g. NESI, SWSI and NSI) and Northland (NNI1 and NNI2) (Fig. 2A, 3A, 4).
This lack of support for any taxonomic subdivision and recognition of geographic structure in Leptospermum is similar to that of Kunzea ericoides (Heenan et al. 2022, 2023a). The basis for species recognition in Kunzea was discussed in some detail by Heenan et al. (2022), with recognition of a single metapopulation lineage (sensu de Queiroz (2007)) referred to as K. ericoides and many of the points raised there (e.g., ecotypic and phenotypic differentiation, reproductive isolation, genotypic variation and gene flow) apply to L. scoparium.
Phylogeography
The genetic structure recovered within NZ L. scoparium comprises a strong north–south geographic pattern within which there is differentiation into regional genetic clusters (Buys et al. 2019; Koot et al. 2022); this study). This geographic structure is explained by environmental variables such as mean annual temperature, potential evapotranspiration ratio and mean temperature of the coldest quarter, which often vary across NZ in steep longitudinal, latitudinal and elevational gradients (Heenan et al. 2021). With the environmental variables providing considerable explanatory power these are considered the major driver of phenotypic and ecotypic variation.
Buys et al. (2019) recognised three geographically structured groups and observed that the NZ Leptospermum clade is characterised by short and often poorly supported branches and that there was a north–south geographic structure. They specifically noted ‘the genetic structure of NZ L. scoparium more strongly reflects geographical proximity than it does morphological similarity’. In the present study, we found a significant relationship between genetic and geographic distance (IBD), and this was further supported by L. repo and the seven NZ regional genetic clusters each having their lowest pairwise FST values with an adjacent region, suggesting L. repo and each of the regional clusters are poorly differentiated and have recurrent gene flow with neighbouring populations. Indicative of this, for example, the East Cape cluster (ECNI) is nested within the central and southern North Island (CSNI) cluster (Fig. 4; pairwise FST 0.1) and the three South Island clusters (pairwise FST 0.06–0.09) are very closely related. Collectively, the three South Island groups comprise a small subset of the genetic variability across all of NZ, are tightly clustered with short edges, and likely reflect repeated genetic bottle-necking through recurring Pleistocene glacial cycles when the woody flora was extirpated or at best patchily distributed.
The overall north to south, landscape-scale, latitudinal distribution with regional genetic clusters that are poorly differentiated (and closely related) in L. scoparium mirrors another microphyllous endemic Myrtaceae, Kunzea ericoides (A.Rich.) Joy Thomps., which is interpreted as comprising a single species and panmictic, metapopulation lineage (Heenan et al. 2022, 2023a). Genetic variation and phylogeography in both K. ericoides and L. scoparium can mostly be explained by variation in environmental variables (Heenan et al. 2023a; this study). Moreover, review of population genetic studies of NZ tree species found latitudinal and regional patterns of genetic variation are common and these often match established biogeographic boundaries for the entire flora (Heenan et al. 2023b).
Genotyping in L. scoparium using a SNP array
A total of 2069 high quality SNPs covering the entire genome was used to genotype L. scoparium and L. repo samples. The SNP array method is attractive compared with whole genome pooled sequencing as it requires less bioinformatics capacity for analysing the data and is cheaper than whole genome sequencing. One key challenge is to obtain a suitable quantity and quality of DNA; however, that problem is the same for whole genome sequencing. A lower success rate was obtained for the Tasmanian samples, which is possibly due to the mediocre quality of the leaf or DNA samples. Most likely the Tasmanian samples may have degraded in the processing stages between sampling from the trees to the DNA extraction and SNP array analysis. The samples extracted in NZ were collected in 2-mL screw cap tubes containing silica beads allowing the sample to desiccate, and were purified individually. The DNA yield obtained using this technique was high (> 20 ng/µL concentration and > 2 µg per sample). Conversely, the Tasmanian samples were collected in 96-well plates, which were kept in a box containing silica beads during the collection, shipped to the lab and extracted in 96-well format. It’s possible that the desiccation process may have failed and that only a subset of 86 samples desiccated properly. The 86 Tasmanian samples successfully genotyped were from seven locations in Tasmania and were therefore used for the genetic diversity analysis. These 86 samples grouped into two gene clusters according to their geographical origins in Tasmania, indicating that our sampling captured genetic diversity within this region.
Conclusion
Our analysis of over 500 L. scoparium samples from Tasmania and NZ screened using over 2000 DNA markers distributed across the genome supports strong genetic differentiation between both regions, and strongly suggests that taxonomic revision of the Australian plants is required. Our results support mānuka as a single endemic NZ species with marked geographic provenances that have significant gene flow, with phenotypic variation largely explained by environmental conditions. These results have significant cultural and commercial implications. The common name in NZ is mānuka, a Te Reo Māori word and taonga (treasure), i.e., issued from the language of NZ indigenous Māori peoples. L. scoparium growing in NZ is strongly differentiated genetically from Australian L. scoparium to the point that we recommend it being taxonomically assigned as a different species. The Te Reo Māori term mānuka (or manuka) should not be used when referring to Australian Leptospermum species. Consequently, our research indicates that honeys marketed from L. scoparium growing in Australia should not be called by the Māori term mānuka.
Data availability
Permission was granted by Māori landowners to re-use the reference genome of L. scoparium ‘Crimson Glory’ (Thrimawithana et al. 2019), as well as the pool-sequencing data and DNA samples of Koot et al. (2022), for the purpose of analysing the genetic difference between NZ and Australia L. scoparium. Further studies using this material, raw sequencing data and genome assembly will require consent from the Māori iwi (tribes) who exercise guardianship for this material according to Aotearoa New Zealand’s Treaty of Waitangi and the international Nagoya protocol on the rights of indigenous peoples. Access to raw and analysed data will require permission from representatives of the iwi.
References
Allan HH (1961) Flora of New Zealand. In: Indigenous tracheophyta: psilopsida, lycopsida, filicopsida, gymnospermae, dicotyledones, vol I. Government Printer, Wellington
Buys MH, Winkworth RC, de Lange PJ, Wilson PG, Mitchell N, Lemmon AR, Moriarty Lemmon E, Holland S, Cherry JR, Klápště J (2019) The phylogenomics of diversification on an island: applying anchored hybrid enrichment to New Zealand Leptospermum scoparium (Myrtaceae). Bot J Linn Soc 191:1–17
Caye K, Deist TM, Martins H, Michel O, François O (2016) TESS3: fast inference of spatial population structure and genome scans for selection. Mol Ecol Resour 16:540–548
Cockayne L (1917) Notes on New Zealand floristic botany, including descriptions of new species, & c. (No. 2). Transactions and Proceedings of the New Zealand Institute 59:56–65
de Lange P, Schmid LM (2021) Leptospermum repo (Myrtaceae), a new species from northern Aotearoa/New Zealand peat bog habitats, segregated from Leptospermum scoparium s. l. Ukr Bot J 78(4):247–265
de Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56:879–886
Dixon P (2003) VEGAN, a package of R functions for community ecology. J Veg Sci 14:927–930
Dray S, Dufour A-B (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20
Fitak RR (2021) OptM: estimating the optimal number of migration edges on population trees using Treemix. Biol Methods Protoc 6(1):bpab017
Frankham R, Ballou SEJD, Briscoe DA, Ballou JD (2002) Introduction to conservation genetics. Cambridge University Press
Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929
Futschik A, Schlötterer C (2010) The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics 186:207–218
Hartl DL, Clark AG, Clark AG (1997) Principles of population genetics, vol 116. Sinauer Associates, Sunderland
Heenan PB, McGlone MS, Mitchell CM, Cheeseman DF, Houliston GJ (2021) Genetic variation reveals broadscale biogeographic patterns and challenges species’ classification in the Kunzea ericoides (kānuka; Myrtaceae) complex from New Zealand. N Z J Bot 59(1):1–25
Heenan PB, McGlone MS, Mitchell CM, Cheeseman DF, Houliston GJ (2022) Genetic variation reveals broad-scale biogeographic patterns and challenges species’ classification in the Kunzea ericoides (kānuka; Myrtaceae) complex from New Zealand. NZ J Bot 60:2–26
Heenan PB, McGlone MS, Mitchell CM, McCarthy JK, Houliston GJ (2023a) Genotypic variation, phylogeography, unified species concept, and the ‘grey zone’ of taxonomic uncertainty in kānuka:recognition of Kunzea ericoides (A.Rich.) Joy Thomps. sens. lat. (Myrtaceae). N Z J Bot 61:1–30
Heenan PB, Lee WG, McGlone MS, McCarthy JK, Mitchell CM, Larcombe MJ, Houliston GJ (2023b) Ecosourcing for resilience in a changing environment. N Z J Bot. https://doi.org/10.1080/0028825X.2023.2210289
Huson DH, Bryant D (2005) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Koot E, Arnst E, Taane M, Goldsmith K, Thrimawithana A, Reihana K, González-Martínez SC, Goldsmith V, Houliston G, Chagné D (2022) Genome-wide patterns of genetic diversity, population structure and demographic history in mānuka (Leptospermum scoparium) growing on indigenous Māori land. Hortic Res 9:uhab012
Li X, Prebble J, de Lange P, Raine J, Newstrom-Lloyd L (2022) Discrimination of pollen of New Zealand mānuka (Leptospermum scoparium agg.) and kānuka (Kunzea spp.)(Myrtaceae). PloS One 17:e0269361
Martinez Arbizu P (2020) pairwiseAdonis: Pairwise multilevel comparison using adonis. R package version 0:4 https://github.com/pmartinezarbizu/pairwiseAdonis
McCarthy JK, Leathwick JR, Roudier P, Barringer JR, Etherington TR, Morgan FJ, Odgers NP, Price RH, Wiser SK, Richardson SJ (2021) New Zealand Environmental Data Stack (NZEnvDS). N Z J Ecol 45:1–8
Mijangos JL, Gruber B, Berry O, Pacioni C, Georges A (2022) dartR v2: an accessible genetic analysis platform for conservation, ecology and agriculture. Methods Ecol Evol 13:2150–2158
Ministry for Primary Industries (2018) Apiculture: Ministry for Primary Industries 2018 apiculture monitoring programme. Wellington, New Zealand https://www.mpi.govt.nz/dmsdocument/34329-apiculture-moniotoring-report-2018
Montanari S, Deng C, Koot E, Bassil NV, Zurn JD, Morrison-Whittle P, Worthington ML, Aryal R, Ashrafi H, Pradelles J (2022) A multiplexed plant-animal SNP array for selective breeding and species conservation applications. bioRxiv bioRxiv 2022.09.07.507051. https://doi.org/10.1101/2022.09.07.507051
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Pembleton LW, Cogan NO, Forster JW (2013) St AMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol Ecol Resour 13:946–952
Perry NB, Brennan NJ, Van Klink JW, Harris W, Douglas MH, McGimpsey JA, Smallfield BM, Anderson RE (1997) Essential oils from New Zealand manuka and kanuka: chemotaxonomy of Leptospermum. Phytochemistry 44:1485–1494
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8:e1002967
Ronghua Y, Mark A, Wilson J (1984) Aspects of the ecology of the indigenous shrub Leptospermum scoparium (Myrtaceae) in New Zealand. NZ J Bot 22:483–507
Schlötterer C, Tobler R, Kofler R, Nolte V (2014) Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat Rev Genet 15:749
Thompson J (1983) Redefinitions and nomenclatural changes within the Leptospermum suballiance of Myrtaceae. Telopea 2:379–383
Thrimawithana AH, Jones D, Hilario E, Grierson E, Ngo HM, Liachko I, Sullivan S, Bilton TP, Jacobs JME, Bicknell R, David C, Deng C, Nieuwenhuizen N, Lopez-Girona E, Tobias P, Morgan E, Perry NB, Lewis DH, Crowhurst RN et al (2019) A whole genome assembly of Leptospermum scoparium (Myrtaceae) for mānuka research. N Z J Crop Hortic Sci 47(4):233–260. https://doi.org/10.1080/01140671.2019.1657911
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370
Williams S, King J, Revell M, Manley-Harris M, Balks M, Janusch F, Kiefer M, Clearwater M, Brooks P, Dawson M (2014) Regional, annual, and individual variations in the dihydroxyacetone content of the nectar of manuka (Leptospermum scoparium) in New Zealand. J Agric Food Chem 62:10332–10340
Acknowledgements
We thank the Honey Landscape governance group and Māori landowners who contributed plant material, Gary Houliston (Manaaki Whenua Landcare Research), Kristen Kohere-Soutar, Tony Wright and Terry Braggins (Te Pitau Ltd) for project planning and funding, Fong Ki Lam (Intertek Australia) for DNA extraction of Tasmanian samples, Tony Fist (Fist Consulting) for sampling in Tasmania, Nico Bordes (Plant & Food Research) for project management, Julien Pradelles (Labogena) for the SNP genotyping and Peter de Lange for the collection of some North Island samples. We thank Dr John Van Klink, Ms Melissa Taane and Mr Ed Morgan for the useful comments on the manuscript. We thank Te Pitau Ltd, Plant & Food Research and the Department of Conservation Science and Research for funding.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by: J. Beaulieu
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chagné, D., Montanari, S., Kirk, C. et al. Single nucleotide polymorphism analysis in Leptospermum scoparium (Myrtaceae) supports two highly differentiated endemic species in Aotearoa New Zealand and Australia. Tree Genetics & Genomes 19, 31 (2023). https://doi.org/10.1007/s11295-023-01606-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01606-w