
Abstract
The evolutionary trajectory of a population both influences and is influenced by characteristics of its genome. A disjunct population, for example is likely to exhibit genomic features distinct from those of continuous populations, reflecting its specific evolutionary history and influencing future recombination outcomes. We examined genetic diversity, population differentiation and linkage disequilibrium (LD) across the highly disjunct native range of the Australian forest tree Eucalyptus globulus, using 203,337 SNPs genotyped in 136 trees spanning seven races. We found support for four broad genetic groups, with moderate F ST, high allelic diversity and genome-wide LD decaying to an r 2 of 0.2 within 4 kb on average. These results are broadly similar to those reported previously in Eucalyptus species and support the ‘ring’ model of migration proposed for E. globulus. However, two of the races (Otways and South-eastern Tasmania) exhibited a much slower decay of LD with physical distance than the others and were also the most differentiated and least diverse, which may reflect the effects of selective sweeps and/or genetic bottlenecks experienced in their evolutionary history. We also show that F ST and rates of LD vary within and between chromosomes across all races, suggestive of recombination outcomes influenced by genomic features, hybridization or selection. The results obtained from studying this species serve to illustrate the genomic effects of population disjunction and further contribute to the characterisation of genomes of woody genera.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genetic differentiation of populations is the basis of evolutionary divergence and speciation, and a key focus of research in evolutionary biology (Hufford and Mazer 2003). Most studies focus on changes in allele frequencies associated with population divergence, but there are also other population parameters which change (Bohonak 1999; Metcalf and Pavard 2007) and can in turn influence their evolutionary trajectories (Trickett and Butlin 1994). Such parameters include genetic diversity, heterozygosity and linkage disequilibrium (LD), all of which define the recombination landscape of a population (Backström et al. 2010; Booker et al. 2017). This landscape may vary not only between populations but also within the genome (Sperisen et al. 2001; Colonna et al. 2014; Gion et al. 2016; Phillips et al. 2018; Soltis and Soltis 2021). The resolution at which such genome-wide variation can now be studied has greatly improved with advances in DNA sequencing and the availability of genome assemblies (Phillips et al. 2018; Soltis and Soltis 2021). These advances have opened the path to more detailed understanding of population and genome variation in LD, which can also be practically applied, for example in genomic selection and genome-wide association genetic studies (GWAS) to inform the density of markers required (Goddard and Hayes 2007; Slatkin 2008; Neale and Kremer 2011).
The non-random association of alleles at different loci, as measured by LD, typically decays with increasing physical separation between loci, and variation in this distance is a population parameter that can influence future recombination outcomes (Slatkin 2008). LD can be studied at various scales, from within genes up to genome-wide averages, with the scale chosen resulting in different aspects of population history and evolution being revealed (Mueller 2004). For instance, examining LD decay distance within a gene can provide insights into the variability inherent in that gene and the associated local rate of recombination and gene conversion (McVean et al. 2004; Xing et al. 2007; Slavov et al. 2012). Alternatively, as LD is influenced by the interplay of gene flow, selection and drift (Slatkin 2008), genome-wide LD can be used to infer population history events. For example, the detection of LD differences between populations can be indicative of past genetic bottlenecks or hybridisation events experienced by one population and not another (Zhang et al. 2004; Noonan et al. 2006). Notable examples of this have been found in human studies, with variable LD observed in different populations thought to have arisen from genetic bottlenecks arising from founder effects (Park 2019). These bottlenecks often have a broad-scale effect on the genome, creating a genome-wide LD signature (Slatkin 2008). In contrast, selective sweeps may cause localised increases in LD around relatively recent advantageous polymorphisms due to insufficient time for recombination to occur (Kim et al. 2007). Localised differences in LD among populations may also arise through variation in chromosomal architecture such as inversions and translocations (Faria et al. 2019; Mostert-O'Neill et al. 2021).
The breeding system of a species will heavily influence the extent of population differentiation. The novel selection pressures faced by crops, for instance create distinctive signatures of extensive LD between different gene pools and populations (Hamblin et al. 2011), which has been observed in many species under domestication (Caldwell et al. 2006; Mather et al. 2007; Chen et al. 2012). While population differences in LD have been observed in relatively few plant species in the wild, some of which include the herb Arabidopsis thaliana (Ostrowski et al. 2006) and the forest tree Populus trichocarpa (Slavov et al. 2012), these impacts are still prevalent. Most forest tree species are outcrossing, leading to high genetic diversity and rapid decay of LD compared to many other plant species (Neale and Kremer 2011; Plomion et al. 2016). Broadly similar genome-wide average LD decay distances ranging from 3 to 6 kb have been observed in tree genera such as Populus (Slavov et al. 2012) and Eucalyptus (Silva-Junior and Grattapaglia 2015) in studies employing 21–30 k markers, although studies in very short regions or employing more dense marker sets have revealed major drops in LD occur within 16–900 bp (Eckert et al. 2009; Arumugasundaram et al. 2011; Marroni et al. 2011; Lu et al. 2016; Murray et al. 2019) (see Discussion).
Belonging to the family Myrtaceae, Eucalyptus is a genus of economically and ecologically important forest trees native to Australia and islands to its north, but grown all over the world for pulp, charcoal and solid wood (Doughty 2000). Eucalyptus globulus, native to south-eastern Australia (Dutkowski and Potts 1999), is one of the major hardwood species grown in pulpwood plantations in temperate regions of the world (Potts et al. 2004; Downham and Gavran 2020; Tomé et al. 2021). A predominantly outcrossing species (Hardner and Potts 1995; Mimura et al. 2009), E. globulus has been classified into 13 races based on distribution and divergence in quantitative genetic traits (Dutkowski and Potts 1999). Genetic structure studies using microsatellite markers have postulated five broad genetic groupings of the races (Yeoh et al. 2012). However, there is only moderate neutral marker differentiation between these races (F ST = 0.09) despite large disjunctions across the species’ natural range, consistent with ongoing, long-distance pollen dispersal (Steane et al. 2006; Mimura et al. 2009; Yeoh et al. 2012). The effect of gene flow is superimposed on a complex history, with evidence for ancient hybridization (McKinnon et al. 2001, 2004b), and environmental adaptation at multiple scales (James and Bell 2000; Foster et al. 2007; Dutkowski and Potts 2012; Freeman et al. 2018; Costa e Silva et al. 2020). Previous studies have used low-density markers to examine population genetic structure (Steane et al. 2006; Yeoh et al. 2012; Costa e Silva et al. 2017) and coarse, genome-wide LD decay (Cappa et al. 2013; Durán et al. 2017) in E. globulus, but access to a more extensive set of markers would allow finer-scale characterisation of genetic structure and differentiation across the genome, and permits comparisons of patterns of LD decay distance.
To this end, this study aims to elucidate the population genetic structure and genome characteristics of E. globulus using a high density set of molecular markers. We examine how LD decay and genomic diversity vary among the different races of E. globulus as well as within and among chromosomes. We also identify genomic regions contributing disproportionately to race and chromosome differentiation. We hypothesise that the various races will exhibit different signatures of LD and diversity at the genome-wide level due to their different evolutionary histories.
Methods
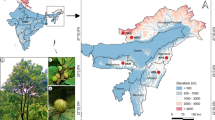
An E. globulus provenance-progeny trial, planted in 1989 near Latrobe in Tasmania, Australia, by Gunns Ltd (now Forico Pty Ltd) was selected for sampling. The trial was established with 570 open-pollinated E. globulus families collected from wild mother trees, with each family represented by a two-tree plot, in each of five replicates. Trees were sampled from this trial within the races of Furneaux (FURN), King Island (KI), West Coast (WC), North-eastern Tasmania (NETAS), South-eastern Tasmania (SETAS) and Southern Tasmania (STAS), along with combined sampling from Gippsland and the Strzelecki Ranges (GIPP), and Western and Eastern Otways (OT) (Fig. 1), until approximately 20 trees from different families had been sampled per race or race combination (hereafter simply referred to as race) (Thavamanikumar et al. 2014). DNA was extracted from cambial scrapings as described in Tibbits et al. (2006).

Geographical distribution of Eucalyptus globulus mother trees sampled from each race in the genetic trial. The grey area indicates the natural distribution of E. globulus. The number in brackets indicates the number of OP progeny sourced from different families from each locality. Mother trees were only represented once per race
Genotyping by whole genome shotgun sequencing
Genomic libraries were prepared using Nextera DNA Library Preparation Kit (Illumina). Double-stranded genomic DNA was used as input for the Nextera ‘tagmentation’ reaction (DNA fragmentation and tagging). The reactions were performed as per the manufacturer’s protocol, replacing the column clean-up (disassociation) step with a 0.6% SDS (sodium dodecyl sulphate) treatment. Library amplification was performed using Nextera Index 1 (i7) and Index 2 (i5) adapters on a Stratagene Mx3005P™ qPCR system (Agilent Technologies), with a thermal profile as follows: 72 °C for 3 min; 98 °C for 30 s; and 5 cycles of 98 °C for 10 s, 63 °C for 30 s and 72 °C for 3 min. SYBR Green I (Thermo Fisher Scientific) in 1:1000 dilution was added to the PCR mixtures for monitoring the amplification process. Amplified libraries were pooled and purified with MinElute PCR Purification Kit (Qiagen) or AMPure XP SPRI (Solid Phase Reversible Immobilisation) paramagnetic beads (Agencourt, Beckman Coulter). Purified library pools were then subjected to size selection using gel electrophoresis by excising the DNA fragments within the desired size range (300–450 bp). The excised libraries were extracted from the gel with QIAquick Gel Extraction Kit (Qiagen) prior to quality assessment with High Sensitivity D1000 ScreenTape using a 2200 TapeStation system (Agilent Technologies). Once assessed, libraries were sequenced using a HiSeq 2000 Sequencing System (Illumina) at the Victorian Government Department of Economic Development, Jobs, Transport and Resources (DEDJTR), Centre for AgriBioscience (AgriBio), Bundoora.
Alignment to a draft genome assembly and SNP calling
A draft genome assembly for E. globulus (X46 v33) was generated from whole genome Illumina paired-end, mate-pair and synthetic long reads from an E. globulus clone (X46) sequenced in a previous project (Bayly et al. 2013; Myburg et al. 2014), and was reference-initiated through alignment to the E. grandis reference genome (Myburg et al. 2014). The Vision software suite (Gydle Inc.) was applied in rounds of iterative automated resolution steps with interactive consensus editing, assembly visualisation and manual curation of end junction sequences to generate the assembly version used in this work. The reference used to initiate the process is not assumed to have the correct linear order and the resolving process specifically detects rearrangements and inversions breaking and re-joining the assembly where conflicts with the raw data are detected. The final draft assembly comprised 13,674 contigs, had a total length of 530.65 Mbp and a contig N50 of 84.7 kbp. While the final assembly was not scaffolded, genome structure is known to be highly conserved between E. globulus and E. grandis (Hudson et al. 2012), so contigs were anchored to the E. grandis reference genome (Myburg et al. 2014) based on comparable sequence to allow contigs to be assigned to one of the 11 chromosomes. A total of 1,162 contigs with length > 100 kbp representing 245 Mbp of the genome were used in this study (see below).
The raw sequence reads from the sampled E. globulus mother trees were quality filtered and aligned using Nuclear v3.2.6 (Gydle Inc., Québec, Canada) to the draft E. globulus genome assembly (X46 v33). SNP variants were discovered using find-snp v2.6.16 (Gydle Inc.). Variants were output in VCF (Variant Call Format) v.4.2 format and filtered or manipulated using VCFtools v0.1.15 (Danecek et al. 2011).
SNP filtering and analysis
From an initial set of 5,594,130 SNPs, data were filtered using VCFtools v0.1.15 (Danecek et al. 2011), initially to SNPs residing on contigs of size > 100 kbp, and individuals to those with < 15% missing data. SNPs were then filtered for quality, with SNPs exhibiting > 10% missing data, a minor allele frequency < 0.2, a minimum read depth < 4 or non-biallelic SNPs removed. To minimise the impact of genotyping errors and paralogous loci, SNPs that deviated from HWE in an extreme manner (i.e. p < 1 × 10−7) were also removed (Pavan et al. 2020). Finally, contigs with < 30 SNPs were removed, resulting in a set of 203,337 SNPs across 1,162 contigs for LD and F ST analysis (see below). The relatively low minimum read depth threshold of 4 was used to achieve the target of at least 30 SNPs per contig examined (for a minimum of 435 SNP pair comparisons). With this relaxed threshold, there was a risk of introducing bias towards homozygotes due to allelic dropout and therefore inflating LD estimates (Nielsen et al. 2011), so average read depth of each race was computed to compare with estimates of LD. The minor allele frequency threshold of 0.2, which is conservative compared to other LD studies (Slavov et al. 2012; Silva-Junior and Grattapaglia 2015), was also adopted to help compensate for the low read depth (in places) of the sequencing data and limit the impact of genotyping errors. Generally lower coverage and a high proportion of missing data resulted in the West Coast race being entirely excluded based on the above criteria.
Population structure between the races was examined using both Admixture v1.3 (Alexander et al. 2009) and fastStructure v1.0 (Raj et al. 2014), using a subset of the above markers. This subset was generated by further filtering to a more stringent missing data rate < 6% and pruning marker pairs in LD (r 2 > 0.2), resulting in a subset of 19,907 unlinked SNPs. In both algorithms (using default parameters), three independent runs were performed for each value of k between 2 and 8. The most probable value of k was defined by cross-validation error in Admixture, or maximised likelihood in fastStructure. The Admixture analysis was also repeated for each chromosome separately once the most probable value of k was selected. Results were visualised using R (R Core Team 2019), with the colour-blind-friendly palette generated using the package viridis (Garnier 2017).
To complement the results from the structure analysis, a principal component analysis (PCA) was undertaken using the same subset of SNPs and individuals. Population differentiation was also examined with F ST using the same subset of markers and the package hierfstat (Jombart 2015). Pairwise F ST were also calculated for each chromosome separately, weighted using the Weir and Cockerham (1984) estimator. To visualise the pairwise results, unweighted pair group method with arithmetic mean (UPGMA) trees were generated using the package phangorn (Schliep 2011). To examine areas of the genome that were contributing to the differentiation, global F ST (i.e. between all pairwise combination of races) was calculated for every contig using the sliding window approach implemented in VCFtools v0.1.15 (Danecek et al. 2011) with the window size set to span entire contigs. These contigs identified to have elevated F ST values were aligned to the E. grandis reference genome, and annotated genes in these regions were tested for enrichment of GO terms using the R package SNP2GO (Szkiba et al. 2014).
Pairwise estimates of linkage disequilibrium (LD) measured by the squared correlation of allele frequencies (r 2) were obtained across every contig, both within races and across races (global estimates) using PLINK v1.9 (Chang et al. 2015) and the set of 203,337 SNPs. A decay curve was fitted for each contig using a nonlinear regression of pairwise LD based on the expectation of r 2 for drift-recombination equilibrium (Hill and Weir 1988) using an R script created by Marroni et al. (2011). As numerous thresholds for measuring LD exist in the literature, we explored the impact of three different thresholds. The first two thresholds, used in other eucalypt studies, employed the decay curve to measure average base pair distance to both a standard r 2 decay threshold of 0.2 and to half-maximum decay (Silva-Junior and Grattapaglia 2015; Murray et al. 2019; Otyama et al. 2019). The third threshold was set to the background LD level within the dataset (Falush et al. 2003; Mather et al. 2007). Background LD was established by selecting five random contigs assigned to each of the 11 chromosomes and calculating r 2 between all SNP pairs possible (excluding those within contig). The upper 95th percentile of these values was set as the background LD threshold. Using the individual contig r 2 estimates, average decay distance and standard errors were calculated at both the race and chromosome level, weighting for contig length and excluding those with curves that failed to reach convergence. Chromosome level comparisons were based on the global r 2 estimates for each contig, whereas race level comparisons were based on r 2 values estimated within each race for each contig, regardless of chromosome. Significant differences in decay distance between races and chromosomes were tested using 1-way ANOVA and Tukey’s HSD test, with the variation among contigs within either factor used as the error in each analysis. Potential patterns of short-range LD were explored by averaging the r 2 values between pairs of SNPs within 5 kb, divided into bins of 100-bp separation (i.e. average r 2 for SNPs separated by 1–100 bp, 101–200 bp etc.). This averaging of the r 2 values among the proximal pairs of SNPs ignored their contig of origin. Genetic diversity was measured using observed and expected heterozygosity and nucleotide diversity (π; setting window size equal to contig size) across contigs using PLINK v1.9, for comparison with the average distance to LD decay at the range-wide, race and chromosome (across race) levels, and tested for significance using ANOVA. To contrast population patterns of selection, Tajima’s D was also calculated at these levels using VCFtools. All results were visualised using the package ggplot (Wickham 2016) in R (R Core Team 2019).
Average LD distance of all contigs (weighted for contig length) were plotted according to their position on the 11 chromosomes. Curves were fitted to the data using locally weighted scatterplot smoothing (LOESS) polynomial regression with a smoothing parameter of 0.1 using R (R Core Team 2019). Peaks in LD were examined for significance by first identifying contigs that were above the 95th percentile in LD decay distance. To test that the clustering of these high LD contigs along the chromosome was greater than expected by chance alone, the position of every contig was randomised across the chromosome. Ten windows along the chromosome were delimited to include 10% of total contigs in each window, and the number of high LD contigs in each window was recorded. This randomisation was repeated 1,000 times. Peaks (within windows) containing a greater number of high LD contigs than the upper limit of the frequency distribution obtained by this bootstrapping were determined to be significant (p < 0.001). This process was repeated for each chromosome. Peaks in expected heterozygosity were also examined in this manner.
Results
Population structure analysis
After filtering SNPs to the criteria detailed in the Methods, 1,162 contigs with 203,337 SNPs were selected for use in LD analyses across 136 individuals. The average contig was 209,837 bp in length, with an average of 175 SNPs per contig spaced at an average of every 1,127 bp. Using a subset of 19,907 unlinked markers, population structure was examined using Admixture and fastStructure, fitting values of k from 2 to 8 (Fig. 2). Both algorithms found k = 4 to be the best at explaining the structure in the data, as it maximised the maximum likelihood values and showed the lowest cross-validation error in fastStructure and Admixture, respectively (Online Resource 1). At k = 4, there was a strong geographic influence on clustering as expected, broadly representing genotypes originating from the following: (1) Tasmania (STAS, SETAS and NETAS; purple group in Fig. 2); (2) the Furneaux (FURN) group of islands (green); (3) southern Victoria (Eastern Otways and Western Otways (OT), Strzelecki Ranges and Gippsland (GIPP); yellow); and (4) King Island (KI; dark blue). Most individuals share some level of genetic cluster identity with individuals from geographically neighbouring races, with the exception of FURN which shows very little admixture with GIPP. The apparent lack of admixture between SETAS and other races is also notable. These observations were also mirrored in a PCA on the same data (Online Resource 2).

Clustering of Eucalyptus globulus trees into different genetic groups using (a) Admixture and (b) fastStructure. Results are shown for values of k from 2 to 8. Each vertical line represents one genotype (an open pollinated progeny from a wild tree), and colours represent partitioning of the genotype to each cluster. Genotypes are grouped by race, including Furneaux (FURN), King Island (KI), North-eastern Tasmania (NETAS), South-eastern Tasmania (SETAS) and Southern Tasmania (STAS), with combined groupings of Gippsland with Strzelecki Ranges (GIPP), and Western Otways with Eastern Otways (OT)
The programs fastStructure and Admixture differed in the stability of genetic groupings past k = 4. The results from fastStructure consistently showed five major genetic groupings from k = 5–8 (with some instability in some genotypes at k = 8) with a distinct separation of GIPP and OT and a stable southern Tasmania group. Admixture was more variable, with NETAS and STAS becoming more distinct from SETAS at k = 5, and GIPP and OT separating at k = 6. Admixture also suggested a less homogenous KI clustering. Concerning the two combined groupings (GIPP and OT), the Strzelecki Ranges race did not separate from Gippsland at any values of k with either algorithm while the Western Otways and Eastern Otways races were only separated at k = 8 in Admixture, suggesting that the molecular genetic differences between these races are small and pooling them into single groups is reasonable.
To provide additional support for these groupings, population differentiation was examined using F ST values. The overall F ST value for the species was 0.059, indicating moderate differentiation between the races (Hartl and Clark 1997). Pairwise comparison of F ST showed that the SETAS and OT races were the most differentiated in allele frequencies, closely followed by SETAS and KI (Table 1). Broadly speaking, there was lower race differentiation within than between Victorian and Tasmanian races (Fig. 3), while the Bass Strait island races were somewhat intermediate, with FURN generally closer to Tasmanian races and KI generally closer to Victorian races, which is in agreement with the population structure (e.g. k = 2 in Fig. 2) and principal component (Online Resource 2) analyses. Examination of differentiation between the races (both in population structure and differentiation) one chromosome at a time revealed that the pattern of differentiation among races was also generally stable across chromosomes (not shown).

Unweighted pair group method with arithmetic mean (UPGMA) phylogram of pairwise F ST values between the Eucalyptus globulus races
Examining population F ST across the genome revealed specific regions where F ST levels are elevated, indicative of regions of the genome that contribute highly to the differentiation observed among the races (Fig. 4). Specifically on chromosomes 1, 2, 6, 8, 9, 10 and 11, regions exceeding a conservative F ST value of 0.15 indicated a high level of differentiation (Hartl and Clark 1997). Examining the annotated genes within contigs at these regions of high differentiation in the E. grandis reference genome revealed 107 genes; however, these genes were not enriched for any specific gene ontology term (Online Resource 3).

Sliding window of global F ST across the Eucalyptus globulus genome. Positions are based on contig alignment with the Eucalyptus grandis reference genome. Chromosomes are delimited by vertical broken lines and labelled. Horizontal broken line indicates the F ST threshold for a window of high differentiation
Linkage disequilibrium and diversity
The distance to threshold decay (r 2 = 0.2), half-maximum decay and background decay (r 2 = 0.039) was calculated within all contigs and all samples in a global analysis, with genome-wide LD decaying to the threshold level within 3,974 bp, to half-maximum within 3,086 bp and to the background rate within 30,101 bp (Table 2). Of the approximately 25 million SNP comparisons, 90% fell below r 2 = 0.2, while 50% of most proximal SNPs (median distance 160 bp) also fell below this threshold. At short ranges, the largest reduction in average allele frequency correlation between SNP pairs was moving from distances of 1–100 bp (average r 2 = 0.37, n = 123,446) to 101–200 bp (average r 2 = 0.29, n = 90,373; Fig. 5). Average allele frequency correlation reaches r 2 = 0.2 within 2.2 kb in this graph, indicative of the variance in results obtained from different methodologies (in this case pooling across SNP pairs regardless of contig of origin versus averages of sliding windows or contigs). For a typical contig, it was extremely rare to see pairwise r 2 > 0.2 beyond 20 kb (Online Resource 4a), while for some contigs higher LD values were common up to 100 kb or beyond (Online Resource 4b & 4c). Although genetic indistinguishability (SNPs seemingly in perfect LD over long distances) can be a factor influencing LD estimates (Skelly et al. 2015), examination of contig distributions of LD decay distances typically revealed stable patterns with no long-distance indistinguishability (Online Resource 4).

Average squared allele frequency correlation (r 2) between SNP pairs within 5 kb in Eucalyptus globulus based on global estimates ignoring race of origin. Each bar represents SNP pairs divided into bins of 100-bp distances (1–100 bp, 101–200 bp etc.)
Strong differences were observed amongst races, with average decay distance to threshold level varying at the race level from 4,646 to 12,249 bp (F 6,8071 = 59.9, p < 0.001), to half-maximum varying from 3,064 to 8,816 bp (F 6,8071 = 51.3, p < 0.001) and to background level from 78,163 to 142,824 bp (F 6,8071 = 101.9, p < 0.001; Table 2). Chromosomes also differed significantly, with average global LD decay distance to the threshold level varying from 3,815 to 11,183 bp (F 10,8067 = 19.9, p < 0.001), to half-maximum varying from 1,116 to 7,761 bp (F 10,8067 = 18.7, p < 0.001) and to background decay rate varying from 72,241 to 132,154 bp (F 10,8067 = 25.9, p < 0.001; Table 3). Both chromosome and race had a significant effect on LD, with chromosomes 1 and 10 and races OT and SETAS showing significantly slower average LD decay across all contigs, and chromosome 5 showing the fastest average LD decay (Table 2, Fig. 6). The trends in the differences among chromosomes and among races were the same regardless of the threshold used and thus the commonly reported (Luikart et al. 2019) threshold decay (r 2 = 0.2) was chosen as the LD statistic to explore longer-range LD further.

Average distance to LD decay (r 2 = 0.2) in Eucalyptus globulus in (a) races and (b) chromosomes. Error bars indicate ± standard error. Significant differences indicated by differing letters as determined by Tukey’s HSD test. Race abbreviations are explained in Fig. 2. Chromosome values are global estimates that ignore race of origin
Heterozygosity was also significantly influenced by race (F 6,136 = 27.6, p < 0.001), with range-wide expected heterozygosity (H e) of 0.423 but race H e ranging from 0.408 to 0.436, with the lowest diversity seen in OT, SETAS and KI (Table 2). An excess of heterozygotes was seen in all races, although the significantly lower observed heterozygosity (H o) in OT and SETAS (F 6,136 = 41.2, p < 0.001) made the difference between H o and H e in these races negligible. After comparing the average read depth per race, we found that OT and SETAS had the highest read depth of all samples despite their lower observed heterozygosity, suggesting no bias had been introduced due to allelic dropout caused by the read depth criteria adopted. Tajima’s D was significantly influenced by race (F 6,8127 = 223.5, p < 0.001), with races all exhibiting positive values (which may be upwardly biased due to the minor allele filtering applied) and ordered similarly to expected heterozygosity (Table 2, Table 3), suggesting higher rates of balancing selection or more recent bottlenecks in the more diverse populations. Nucleotide diversity was also significantly influenced by race (F 6,8127 = 17.3, p < 0.001), and ordered similarly to expected heterozygosity, with the exception of OT and SETAS which unexpectedly had slightly higher nucleotide diversity. As nucleotide diversity can be interpreted as the percentage of sites expected to differ between two randomly selected individuals in the population, this pattern appears to be caused by the greater diversity of observed genotypes in these less heterozygous populations. Each population statistic was on average more variable among races than among chromosomes (Table 3).
Characterising LD variation within chromosomes
Mapping LD decay across the genome showed variable decay distance along all chromosomes, with several showing slow LD decay in the central third (Fig. 7). Highly significant clustering (as determined by bootstrapping, p < = 0.001) of contigs with slow LD decay was detected on all chromosomes barring 5, 7 and 11 (Online Resource 5). These peaks in LD decay distance were present (at various intensities) in the same chromosomal region across all races while spanning different contigs in the region, suggestive of common genomic features influencing decay distance. There are several smaller peaks present on other chromosomes (for example those in the first half of chromosome 9) but these were generally associated with areas of slow LD decay unique to the OT and SETAS races. These two races had the highest average genome-wide LD decay distance (Table 2) and, while there were unique peaks, for the most part, their contigs with slower LD decay compared with other races were spread across the genome (Fig. 7). Several of these race-specific contigs had LD decay distances of over 100 kb (> 25 X the global across-race estimate), suggesting large haplotype blocks exist in these genomic regions in these races.

LD decay distance along each Eucalyptus globulus chromosome by race. Each point represents the average distance to LD decay (r 2 = 0.2) for a contig in a race, with chromosome position of the contigs based on synteny with the E. grandis reference genome. Curve drawn using locally weighted scatterplot smoothing (LOESS) polynomial regression with a smoothing parameter of 0.1. Contigs associated with the two races with the highest average distance to LD decay are coloured
Reduced diversity (i.e. expected heterozygosity) is generally associated with extended distance to LD decay (Smith et al. 2005), which we observed broadly at the race level, although not reaching significance (r = − 0.61, p = 0.14; Table 2). When comparing contig-level expected heterozygosity to the chromosome LD peaks, very few genomic regions with reduced diversity featured increased LD (Fig. 8), with the exception of a small number of low-diversity contigs near the LD peak on chromosomes 2 and 10. Bootstrapping of contig positions on chromosome 2 showed the clustering of low diversity contigs in this window associated with the peak in LD decay distance was significant (p < 0.01). Races were relatively consistent in their genetic diversity across chromosomes (with OT and SETAS exhibiting consistently lower diversity). Contig-level observed heterozygosity showed a similar pattern, with OT and SETAS exhibiting much lower heterozygosity overall and a significant (from bootstrapping, p < 0.01) association of slow LD with contigs exhibiting low observed heterozygosity on chromosome 2 (Online Resource 6).

Expected heterozygosity (H e) along each Eucalyptus globulus chromosome. Each point represents a contig position based on synteny with the E. grandis reference genome for each race. The solid curve shows the average expected heterozygosity across all races, drawn using locally weighted scatterplot smoothing (LOESS) polynomial regression with a smoothing parameter of 0.1. The dotted curve shows the smoothed LD decay curve along chromosomes, averaged across all races (see Fig. 6). Contigs associated with the two races with the highest average distance to LD decay are coloured. Red arrows along the x-axis indicate positions of contigs with elevated F ST (see Fig. 4)
Notable findings from a previous study of recombination rate in E. globulus were an unusually strong positive correlation between gene density and recombination rate, and a negative correlation between recombination rate and genetic diversity, both at the chromosome level (Gion et al. 2016). At the chromosome level, there was no significant correlation between gene density (obtained via annotated genes in the E. grandis reference genome) and average LD decay distance (r = 0.625, p > 0.05, Online Resource 7). However, examining gene density versus proportion of contigs with high LD decay distances in ten windows across each chromosome revealed a weak negative correlation (r = − 0.32, p < 0.001, Online Resource 8) which, if assuming high LD as a proxy for lower recombination, would be consistent with Gion et al. (2016).
Discussion
This study contributes to the increasing literature regarding genome-wide characterisation of population characteristics in wild plants (Ostrowski et al. 2006; Slavov et al. 2012; Silva-Junior and Grattapaglia 2015; Murray et al. 2019). Several key points have emerged. First, the genetic structure and degree of differentiation between E. globulus races was largely shaped by the disjunctions in its distribution, as observed in previous microsatellite studies (Steane et al. 2006; Yeoh et al. 2012). Secondly, the genome-wide LD decay distance estimated with our SNP data set varied significantly between the races of E. globulus. Regardless of the LD threshold used, the South-eastern Tasmanian and Otways races exhibited greater than expected differences from their neighbouring races, with significantly greater LD decay distances and lower observed heterozygosity, along with lower outbreeding (F) and expected heterozygosity, likely reflecting their distinct evolutionary histories. Lastly, some genomic regions contributed disproportionately to both differentiation and LD decay distance across all races (Fig. 7), possibly indicating regional genomic features such as repetitive content or chromatin architecture influencing these estimates.
Variation between species
Eucalyptus globulus has a mixed mating system but is predominantly outcrossing (Mimura et al. 2009) and thus would be expected to have reasonably low genome-wide average LD in contrast to plants with higher levels of selfing (Nordborg 2000; Flint-Garcia et al. 2003). Indeed, the global LD decay distance to r 2 = 0.2 averaged across contigs of 4 kb estimated with our genome-wide E. globulus SNP data set is quite low in comparison with annual selfing plant species such as wild soybean (75 kb, Lam et al. 2010), chickpea (450 kb, Bajaj et al. 2015) and mungbean (60 kb, Noble et al. 2018), and perennial outcrossing species such as Arabidopsis lyrata (> 100 kb, Hämälä and Savolainen 2019), but similar to that estimated for other outcrossing tree species such as E. grandis (4–6 kb, Silva-Junior and Grattapaglia 2015) and poplar (3–6 kb, Slavov et al. 2012).
However, direct comparisons of LD decay distance between studies are problematic and affected by factors including variations in methodology, filtering (especially minor allele frequency which can cause LD inflation; Boyles et al. 2005), and marker type and density, as well as the point on the decay curve comparisons are made. Therefore, it is important to consider the scale at which LD is being examined and the conclusions which can validly be drawn from results at the scale examined. For example, the average LD decay distances to r 2 = 0.2 of 3–6 kb for E. grandis and poplar employing 21 k–30 k markers (Slavov et al. 2012; Silva-Junior and Grattapaglia 2015) are in marked contrast with distances to half-maximum decay of 92–113 bp reported in another Eucalyptus study examining LD with millions of SNPs (Murray et al. 2019), which is likely to reflect the different patterns and processes impacting long- and short-range LD, respectively (Andolfatto and Nordborg 1998; Frisse et al. 2001; Pritchard and Przeworski 2001; Tenesa et al. 2007; Slavov et al. 2012). Similarly, even within E. globulus, variable LD decay distances have been reported, no doubt representing different scales of study, marker densities and population characteristics. An early study of SNPs in 12 candidate genes in four native E. globulus populations, for example suggested that LD within genes decayed rapidly to an r 2 = 0.2 within 500 bp (Thavamanikumar et al. 2011), whereas relatively low-density genome-wide studies of E. globulus breeding populations suggest that the distance to reach this average LD threshold is greater (Durán et al. 2017; Ballesta et al. 2018, 2019), and close to our global estimate of 4 kb (although the estimate from one complex breeding population reported r 2 = 0.2 at 21.8 kb (Quezada et al. 2022)). Evidence for the impact of marker density/scale of observation on estimates of LD decay distance comes from our fine-scale examination of global LD decay distance using the subset of markers separated by less than 5 kb, which show much more rapid decay than our contig average estimates. The steepest decline in LD occurred between 100 and 200 bp, consistent with the distance to half-maximum decay reported in Murray et al. (2019). This initial rapid LD decline over short genomic distance no doubt reflects short-range LD, shaped by ancient mutations and recombination events (Tenesa et al. 2007), along with the complicating factor of gene conversion (Andolfatto and Nordborg 1998; Frisse et al. 2001). In contrast, the LD decay threshold of r 2 = 0.2 on which we focused likely reflects a long-range LD more influenced by events in relatively recent population history such as bottlenecks, admixture and selection (Tenesa et al. 2007; Slavov et al. 2012), as well as chromosomal structural features (Vallejo et al. 2018; Todesco et al. 2020; Valenzuela et al. 2021).
Long-lived woody species like forest trees are known for their generally higher diversity compared to other life forms (Hamrick et al. 1992; Broadhurst et al. 2017), and those that outcross like poplar and pine often show higher observed than expected heterozygosity (Lu et al. 2016; Bothwell et al. 2017). An excess of heterozygotes was observed across all E. globulus races, consistent with the outbreeding nature of E. globulus, animal-mediated pollen dispersal (Mimura et al. 2009) and strong-post-dispersal selection against the products of inbreeding (Costa e Silva et al. 2014; Nickolas et al. 2019). While heterozygote excess was not observed in previous studies of E. globulus races (Jones et al. 2006; Steane et al. 2006; Yeoh et al. 2012), this is likely due to the nature of the markers used (all the cited studies used microsatellites), as the presence (Ollitrault et al. 2015), direction and intensity (Ebadi et al. 2019; Zimmerman et al. 2020) of the difference between observed and expected heterozygosity can vary widely between microsatellites and SNPs.
Variation between races
Population structure analyses suggested that the main divergence between E. globulus races is found between the mainland and Tasmania (k = 2), in agreement with previous microsatellite studies in E. globulus (Steane et al. 2006; Yeoh et al. 2012), with the geographically intermediate island races in western and eastern Bass Strait markedly different from each other. Accordingly, the best structure grouping was the four genetic groups: Victoria, King Island, Tasmania and the Furneaux islands. However, our next best grouping in the fastStructure analysis separated the mainland groups Gippsland and Otways consistent with microsatellite studies reported by Yeoh et al. (2012), Costa e Silva et al. (2017) and Thavamanikumar et al. (2014). Indeed, the five-group structure is the most consistent with the disjunctions between the sampled races, i.e. those imposed by Bass Strait and the relatively short disjunction between the Otways and Gippsland races. The pairwise differentiation between races also matched the relationships observed by Callister et al. (2021) (with the exception of the King Island and Otways pairing, possibly due to our pooling of the two eastern and western Otways races). It should be noted that the current study was unable to include the small disjunct western races of E. globulus which, in other analyses, link with the King Island and Otways races (Steane et al. 2006; Yeoh et al. 2012; Callister et al. 2021). In combination with evidence from shared chloroplast haplotypes (which are maternally inherited and therefore dispersed by seed only), this provides further evidence of a western migration route linking the Otways to Tasmania via a land-bridge formed in glacial periods (McKinnon et al. 2001) and now only above water through King Island. Shared chloroplast haplotypes between Gippsland and the Furneaux Islands, but not between these races and Tasmanian races (Freeman et al. 2001) argues for a historic barrier to seed dispersal between the Furneaux Islands and Tasmania. There are also some shared chloroplast haplotypes between Western Tasmania and the races on the east coast of Tasmania. In contrast, this study showed admixture in the nuclear genome between the north-eastern Tasmania and the Furneaux Islands races, consistent with the findings of chemical and microsatellite studies (Wallis et al. 2011; Yeoh et al. 2012). All these patterns can be reconciled by a ring migration model, whereby E. globulus migrated southward by seed to Tasmania from the Otways, onto the west coast of Tasmania, through Southern Tasmania and northward on the east coast of Tasmania but not reaching the Furneaux Islands by seed from Tasmania (Freeman et al. 2001). The Furneaux islands were then likely populated by seed from the Gippsland area, with subsequent pollen-mediated gene flow causing the admixture seen in the nuclear genome between the Furneaux Islands and Tasmania.
While LD decay distance varied among races in E. globulus, the average global LD decay distance for the combined races was less than almost any value obtained for a single race, at all thresholds of r 2 (barring one instance of race half-maximum decay). This is expected, as LD measurements are a function of allele frequencies and thus pooling sub-populations is likely to average any race-specific divergences in allele frequency (Evans and Cardon 2005). Therefore, a species-wide estimate is likely to under- or over-estimate LD decay distance in the presence of population structure, as has been previously reported in Populus trichocarpa (Slavov et al. 2012) and E. grandis (Silva-Junior and Grattapaglia 2015), and will obscure signals of distinct evolutionary histories experienced by sub-populations. For instance, in P. trichocarpa LD decay distance varied from around 2.5 kb in one sub-population up to > 6 kb in several others, with the higher values thought to be caused by bottlenecks or founder effects experienced over the last few hundred generations (Slavov et al. 2012). In the present case, reconciling the slow LD decay and low heterozygosity in the South-eastern Tasmanian and Otways races with their evolutionary history is difficult. These races of E. globulus both coincide with/are adjacent to hypothesised forest glacial refugia (Freeman et al. 2001; McKinnon et al. 2004a; Nevill et al. 2014), and there is evidence of extensive ancient hybridization with other eucalypt species in south-eastern Tasmania (McKinnon et al. 2004b). Expectations would suggest that these refugia should harbour higher than average genetic diversity relative to the other races (Médail and Diadema 2009), but this was not seen in our SNP data. Indeed, the dominant signal in these populations is one associated with higher inbreeding, with almost no heterozygote excess, in contrast to that observed in the other races (Wright 1943). A higher F-statistic can indicate geographic sub-structuring (De Meeûs 2017), which could be argued for the Otways race as in our study it is a combination of the Eastern and Western Otways races. However, this is unlikely due to the homogeneity of the genetic structure in both Otways and South-eastern Tasmania and would not explain their low observed heterozygosity. While evidence from chloroplast sharing suggests that ancient hybridization may have affected the South-eastern Tasmania race, any effect on nuclear genome diversity appears to be small (McKinnon et al. 2004b). Likewise, while more recent hybridization can lead to increases in LD, it is also generally associated with increases in diversity (Fridman 2015; Goicoechea et al. 2015), which was not observed here. Population bottlenecks/founder events (such as post-glacial migration from refugia) occurring post-hybridization could potentially lead to a genome-wide signal of slow LD decay (Finlay et al. 2017), which is consistent with our observations in the Otways and South-eastern Tasmania races. In contrast, the impact of a selective sweep would generally be localised around the advantageous allele rather than genome-wide (Galtier et al. 2000). To this effect, we did observe areas with a coincidence of low diversity and slow LD decay, one on chromosome 2 which was most evident in the Otways race, and one on chromosome 10 most evident in South-eastern Tasmania (Fig. 7). As both genome-wide and localised patterns are present in these two races, it is possible both processes contribute to their slower than average LD decay. Furthermore, given there are also observed peaks in slow LD decay which appear to be specific to these two races (e.g. chromosome 9) that did not coincide with low heterozygosity peaks, the possibility that chromosomal rearrangement variants also contribute to their slower than average LD decay cannot be dismissed (Mostert-O'Neill et al. 2021).
Variation between and within chromosomes
There was distinct variation between chromosomes when race was accounted for, with chromosomes 1 and 10 exhibiting significantly slower LD decay than any other chromosome, and chromosome 5 exhibiting the fastest LD decay. At the broad chromosome-wide scale, differences in recombination rate are likely to be the main driver of differences in LD (Flint-Garcia et al. 2003). Considering broad chromosome characteristics, chromosome length is one factor that is known to influence recombination rate (Shen et al. 2017). Consistent with this observation, chromosomes 1 and 10 are among the shorter and chromosome 5 among the longer chromosomes in E. grandis and have the highest and lowest recombination rates, respectively (Gion et al. 2016). Although this is also likely the case in E. globulus given their known synteny (Hudson et al. 2012), the recombination rates are in the opposite direction as would be predicted by the LD values exhibited in this study (except in the presence of high gene density, see next section). However, chromosomes 4 and 9 are similarly short and chromosomes 3 and 8 are similarly long, all of which exhibit LD decay distances closer to the average. Another possibility contributing to chromosomal differences and some of the isolated peaks in LD is differences in chromosome structural polymorphisms, as reported in E. grandis (Mostert-O'Neill et al. 2021). Looking within chromosomes, large peaks of elevated decay distance were found in similar genomic regions in all races on most chromosomes, suggesting common genomic features influencing LD at these locations. Notably, the largest peaks were found on chromosomes 1 and 10, likely contributing to their significantly higher LD than other chromosomes. Some peaks also coincided with areas that exhibited high levels of F ST, which when taken together is suggestive of key areas of differentiation between the races. Interestingly, a similar pattern of global F ST values (elevated F ST in the same regions of the same chromosomes) has been observed before in a study of six different Eucalyptus species (Hudson et al. 2015). Many of the species-differentiating markers identified in that study align well with peaks in F ST in this study (Online Resource 9), implying a level of conservation in these features across the subgenus. These could represent large adaptive haploblocks (Todesco et al. 2020) caused by recombination-inhibiting structural variation such as inversions (Vallejo et al. 2018). Many of the peaks in LD and F ST were in a broadly central region of the chromosome and may also be indicative of supressed recombination around the centromere. There is cytogenetic evidence for centromere position in E. globulus chromosomes; however, we cannot identify homologous chromosomes between the reference genome and the cytology study (Ribeiro et al. 2016). Other elements of chromosome structure could also play a role, for example polymorphic transposable elements have been shown to elevate LD decay distance (Choudhury et al. 2019), while chromatin architecture has also been associated with changes in LD (Smith et al. 2005; Henderson 2012; Jabbari et al. 2018).
Genomic regions subject to selective sweeps due to adaptive genes will also result in reduced diversity and subsequent slower decay of LD (Kim and Nielsen 2004), which was observed on chromosome 2 in this study. This is more likely in areas of high gene density, as they are generally subject to higher rates of selection and subsequently exhibit lower diversity, such as seen in E. grandis (Silva-Junior and Grattapaglia 2015). Therefore, in regions of high recombination rate along with high gene density, the typical positive correlation between recombination rate and diversity may be reversed by purifying selection, as selection may have increased efficacy due to high recombination rate (Pál et al. 2001). Notably, broad selective sweeps have been shown to leave a signature of slow LD decay (Kim and Nielsen 2004), which would develop despite the high recombination rate as areas become more homogenous (Gion et al. 2016). We found chromosome average LD in E. globulus to be negatively correlated with diversity and positively correlated with gene density, consistent with this hypothesis.
Conclusions
Genetic differentiation was detected between the different races of E. globulus, and their groupings reflected the main disjunction across its distribution. The levels of linkage disequilibrium and diversity in E. globulus were comparable to other genome-wide and range-wide estimates in other outbreeding tree species, including eucalypts. This study emphasises the differences between short-and long-range LD and focuses on contig variation in longer-range decay (r 2 = 0.2), which is shown to vary between races, suggestive of specific evolutionary histories. Differences were also observed within and between chromosomes, suggesting genomic features or processes associated with recombination suppression or diversity reduction. The mechanisms underlying the variation in LD decay in specific areas of the genome require further study and will need to be considered carefully when performing marker-phenotype associations.
Data availability
The read data used to generate the SNPs in this study are accessible on the European Nucleotide Archive (accession PRJEB47881). The E. globulus draft genome assembly (X46 v33) is available on reasonable request to the corresponding author.
Change history
22 July 2022
Handling editor name correction.
References
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Andolfatto P, Nordborg M (1998) The effect of gene conversion on intralocus associations. Genetics 148:1397–1399
Arumugasundaram S, Ghosh M, Veerasamy S, Ramasamy Y (2011) Species discrimination, population structure and linkage disequilibrium in Eucalyptus camaldulensis and Eucalyptus tereticornis using SSR markers. PLoS ONE 6:e28252
Backström N, Forstmeier W, Schielzeth H, Mellenius H, Nam K, Bolund E et al (2010) The recombination landscape of the zebra finch Taeniopygia guttata genome. Genome Res 20:485–495
Bajaj D, Das S, Badoni S, Kumar V, Singh M, Bansal KC et al (2015) Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Sci Rep 5:12468
Ballesta P, Serra N, Guerra FP, Hasbún R, Mora F (2018) Genomic prediction of growth and stem quality traits in Eucalyptus globulus Labill. at its southernmost distribution limit in Chile. Forests 9:779
Ballesta P, Maldonado C, Pérez-Rodríguez P, Mora F (2019) SNP and haplotype-based genomic selection of quantitative traits in Eucalyptus globulus. Plants 8:331
Bayly MJ, Rigault P, Spokevicius A, Ladiges PY, Ades PK, Anderson C et al (2013) Chloroplast genome analysis of Australian eucalypts – Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae). Mol Phylogenet Evol 69:704–716
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74:21–45
Booker TR, Ness RW, Keightley PD (2017) The recombination landscape in wild house mice inferred using population genomic data. Genetics 207:297–309
Bothwell HM, Cushman SA, Woolbright SA, Hersch-Green EI, Evans LM, Whitham TG et al (2017) Conserving threatened riparian ecosystems in the American West: precipitation gradients and river networks drive genetic connectivity and diversity in a foundation riparian tree (Populus angustifolia). Mol Ecol 26:5114–5132
Boyles AL, Scott WK, Martin ER, Schmidt S, Li Y-J, Ashley-Koch A et al (2005) Linkage disequilibrium inflates type I error rates in multipoint linkage analysis when parental genotypes are missing. Hum Hered 59:220–227
Broadhurst L, Breed M, Lowe A, Bragg J, Catullo R, Coates D et al (2017) Genetic diversity and structure of the Australian flora. Divers Distrib 23:41–52
Caldwell KS, Russell J, Langridge P, Powell W (2006) Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics 172:557–567
Callister AN, Bradshaw BP, Elms S, Gillies RAW, Sasse JM, Brawner JT (2021) Single-step genomic BLUP enables joint analysis of disconnected breeding programs: an example with Eucalyptus globulus Labill. G3 Genes|Genomes|Genetics 11
Cappa EP, El-Kassaby YA, Garcia MN, Acuña C, Borralho NMG, Grattapaglia D et al (2013) Impacts of population structure and analytical models in genome-wide association studies of complex traits in forest trees: a case study in Eucalyptus globulus. PLoS ONE 8:e81267
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7
Chen X, Min D, Yasir TA, Hu Y-G (2012) Genetic diversity, population structure and linkage disequilibrium in elite chinese winter wheat investigated with SSR markers. PLoS ONE 7:e44510
Choudhury RR, Rogivue A, Gugerli F, Parisod C (2019) Impact of polymorphic transposable elements on linkage disequilibrium along chromosomes. Mol Ecol 28:1550–1562
Colonna V, Ayub Q, Chen Y, Pagani L, Luisi P, Pybus M et al (2014) Human genomic regions with exceptionally high levels of population differentiation identified from 911 whole-genome sequences. Genome Biol 15:1–14
Costa e Silva J, Vaillancourt RE, Steane DA, Jones RC, Marques C (2017) Microsatellite analysis of population structure in Eucalyptus globulus. Genome 60:770–777
Costa e Silva J, Potts BM, Harrison PA (2020) Population divergence along a genetic line of least resistance in the tree species Eucalyptus globulus. Genes 11:1095
Costa e Silva J, Potts BM, Lopez GA (2014) Heterosis may result in selection favouring the products of long-distance pollen dispersal in Eucalyptus. PLoS ONE 9
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
De Meeûs T (2017) Revisiting FIS, FST, Wahlund effects, and null alleles. J Hered 109:446–456
Doughty RW (2000) The Eucalyptus: a natural and commercial history of the gum tree. Johns Hopkins University Press, Baltimore and London
Downham R, Gavran M (2020) Australian plantation statistics 2020 update. Australian Bureau of Agricultural and Resource Economics and Sciences (ABARES), Canberra
Durán R, Isik F, Zapata-Valenzuela J, Balocchi C, Valenzuela S (2017) Genomic predictions of breeding values in a cloned Eucalyptus globulus population in Chile. Tree Genet Genomes 13:74
Dutkowski GW, Potts BM (1999) Geographic patterns of genetic variation in Eucalyptus globulus ssp. globulus and a revised racial classification. Aust J Bot 47:237–263
Dutkowski GW, Potts BM (2012) Genetic variation in the susceptibility of Eucalyptus globulus to drought damage. Tree Genet Genomes 8:757–773
Ebadi A, Ghaderi N, Vafaee Y (2019) Genetic diversity of Iranian and some European grapes as revealed by nuclear and chloroplast microsatellite and SNP molecular markers. J Hortic Sci Biotechnol 94:599–610
Eckert AJ, Wegrzyn JL, Pande B, Jermstad KD, Lee JM, Liechty JD et al (2009) Multilocus patterns of nucleotide diversity and divergence reveal positive selection at candidate genes related to cold hardiness in coastal Douglas fir (Pseudotsuga menziesii var. menziesii). Genetics 183:289–298
Evans DM, Cardon LR (2005) A comparison of linkage disequilibrium patterns and estimated population recombination rates across multiple populations. Am J Hum Genet 76:681–687
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Faria R, Chaube P, Morales HE, Larsson T, Lemmon AR, Lemmon EM et al (2019) Multiple chromosomal rearrangements in a hybrid zone between Littorina saxatilis ecotypes. Mol Ecol 28:1375–1393
Finlay CMV, Bradley CR, Preston SJ, Provan J (2017) Low genetic diversity and potential inbreeding in an isolated population of alder buckthorn (Frangula alnus) following a founder effect. Sci Rep 7:3010
Flint-Garcia SA, Thornsberry JM, B ES IV (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Foster SA, McKinnon GE, Steane DA, Potts BM, Vaillancourt RE (2007) Parallel evolution of dwarf ecotypes in the forest tree Eucalyptus globulus. New Phytol 175:370–380
Freeman J, Jackson H, Steane D, McKinnon G, Dutkowski G, Potts B et al (2001) Chloroplast DNA phylogeography of Eucalyptus globulus. Aust J Bot 49:585–596
Freeman JS, Hamilton MG, Lee DJ, Pegg GS, Brawner JT, Tilyard PA et al (2018) Comparison of host susceptibility to native and exotic pathogens provides evidence for pathogen imposed selection in forest trees. New Phytol 221:2261–2272
Fridman E (2015) Consequences of hybridization and heterozygosity on plant vigor and phenotypic stability. Plant Sci 232:35–40
Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 69:831–843
Galtier N, Depaulis F, Barton NH (2000) Detecting bottlenecks and selective sweeps from DNA sequence polymorphism. Genetics 155:981–987
Garnier S (2017) viridis: Default color maps from ‘matplotlib’. R package version 040 https://CRAN.R-project.org/package=viridis
Gion J-M, Hudson CJ, Lesur I, Vaillancourt RE, Potts BM, Freeman JS (2016) Genome-wide variation in recombination rate in Eucalyptus. BMC Genomics 17:590
Goddard M, Hayes B (2007) Genomic selection. J Anim Breed Genet 124:323–330
Goicoechea PG, Herrán A, Durand J, Bodénès C, Plomion C, Kremer A (2015) A linkage disequilibrium perspective on the genetic mosaic of speciation in two hybridizing Mediterranean white oaks. Heredity 114:373–386
Hämälä T, Savolainen O (2019) Genomic patterns of local adaptation under gene flow in Arabidopsis lyrata. Mol Biol Evol 36:2557–2571
Hamblin MT, Buckler ES, Jannink J-L (2011) Population genetics of genomics-based crop improvement methods. Trends Genet 27:98–106
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. New Forest 6:95–124
Hardner C, Potts B (1995) Inbreeding depression and changes in variation after selfing in Eucalyptus globulus ssp. globulus. Silvae Genetica 44:46–54
Hartl DL, Clark AG (1997) Principles of population genetics vol 116. Sinauer Associates Sunderland, MA
Henderson IR (2012) Control of meiotic recombination frequency in plant genomes. Curr Opin Plant Biol 15:556–561
Hill WG, Weir BS (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33:54–78
Hingston AB, Gartrell BD, Pinchbeck G (2004) How specialized is the plant–pollinator association between Eucalyptus globulus ssp. globulus and the swift parrot Lathamus discolor? Austral Ecol 29:624–630
Hudson CJ, Kullan ARK, Freeman JS, Faria DA, Grattapaglia D, Kilian A et al (2012) High synteny and colinearity among Eucalyptus genomes revealed by high-density comparative genetic mapping. Tree Genet Genomes 8:339–352
Hudson CJ, Freeman JS, Myburg AA, Potts BM, Vaillancourt RE (2015) Genomic patterns of species diversity and divergence in Eucalyptus. New Phytol 206:1378–1390
Hufford KM, Mazer SJ (2003) Plant ecotypes: genetic differentiation in the age of ecological restoration. Trends Ecol Evol 18:147–155
Jabbari K, Wirtz J, Rauscher M, Wiehe T (2018) Interdependence of linkage disequilibrium, chromatin architecture and compositional genome organization of mammals. bioRxiv:293837
James SA, Bell DT (2000) Influence of light availability on leaf structure and growth of two Eucalyptus globulus ssp. globulus provenances. Tree Physiol 20:1007–1018
Jombart JGaT (2015) hierfstat: Estimation and tests of hierarchical F-statistics. R package version 004–22 https://CRAN.R-project.org/package=hierfstat
Jones TH, Steane DA, Jones RC, Pilbeam D, Vaillancourt RE, Potts BM (2006) Effects of domestication on genetic diversity in Eucalyptus globulus. For Ecol Manage 234:78–84
Kim Y, Nielsen R (2004) Linkage disequilibrium as a signature of selective sweeps. Genetics 167:1513–1524
Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S et al (2007) Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet 39:1151
Lam H-M, Xu X, Liu X, Chen W, Yang G, Wong F-L et al (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053
Lu M, Krutovsky KV, Nelson CD, Koralewski TE, Byram TD, Loopstra CA (2016) Exome genotyping, linkage disequilibrium and population structure in loblolly pine (Pinus taeda L.). BMC Genomics 17:730
Luikart G, Kardos M, Hand BK, Rajora OP, Aitken SN, Hohenlohe PA (2019) Population genomics: advancing understanding of nature. In: Rajora OP (ed) Population genomics: concepts, approaches and applications. Springer International Publishing pp 3–79. https://doi.org/10.1007/13836_2018_60
Marroni F, Pinosio S, Zaina G, Fogolari F, Felice N, Cattonaro F et al (2011) Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genet Genomes 7:1011–1023
Mather KA, Caicedo AL, Polato NR, Olsen KM, McCouch S, Purugganan MD (2007) The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177:2223–2232
McKinnon GE, Vaillancourt RE, Jackson HD, Potts BM (2001) Chloroplast sharing in the Tasmanian eucalypts. Evolution 55:703–711
McKinnon GE, Jordan GJ, Vaillancourt RE, Steane DA, Potts BM (2004) Glacial refugia and reticulate evolution: the case of the Tasmanian eucalypts. Philos Trans R Soc Lond b: Biol Sci 359:275–284
McKinnon GE, Vaillancourt RE, Steane DA, Potts BM (2004) The rare silver gum, Eucalyptus cordata, is leaving its trace in the organellar gene pool of Eucalyptus globulus. Mol Ecol 13:3751–3762
McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584
Médail F, Diadema K (2009) Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr 36:1333–1345
Metcalf CJE, Pavard S (2007) Why evolutionary biologists should be demographers. Trends Ecol Evol 22:205–212
Mimura M, Barbour RC, Potts BM, Vaillancourt RE, Watanabe KN (2009) Comparison of contemporary mating patterns in continuous and fragmented Eucalyptus globulus native forests. Mol Ecol 18:4180–4192
Mostert-O’Neill MM, Reynolds SM, Acosta JJ, Lee DJ, Borevitz JO, Myburg AA (2021) Genomic evidence of introgression and adaptation in a model subtropical tree species, Eucalyptus grandis. Mol Ecol 30:625–638
Mueller JC (2004) Linkage disequilibrium for different scales and applications. Brief Bioinform 5:355–364
Murray KD, Janes JK, Jones A, Bothwell HM, Andrew RL, Borevitz JO (2019) Landscape drivers of genomic diversity and divergence in woodland Eucalyptus. Mol Ecol 28:5232–5247
Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J et al (2014) The genome of Eucalyptus grandis. Nature 510:356–362
Neale DB, Kremer A (2011) Forest tree genomics: growing resources and applications. Nat Rev Genet 12:111
Nevill PG, Després T, Bayly MJ, Bossinger G, Ades PK (2014) Shared phylogeographic patterns and widespread chloroplast haplotype sharing in Eucalyptus species with different ecological tolerances. Tree Genet Genomes 10:1079–1092
Nickolas H, Harrison PA, Tilyard P, Vaillancourt RE, Potts BM (2019) Inbreeding depression and differential maladaptation shape the fitness trajectory of two co-occurring Eucalyptus species. Ann for Sci 76:10
Nielsen R, Paul JS, Albrechtsen A, Song YS (2011) Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet 12:443–451
Noble TJ, Tao Y, Mace ES, Williams B, Jordan DR, Douglas CA et al (2018) Characterization of linkage disequilibrium and population structure in a mungbean diversity panel. Frontiers in Plant Science 8
Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J et al (2006) Sequencing and analysis of Neanderthal genomic DNA. Science 314:1113–1118
Nordborg M (2000) Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154:923–929
Ollitrault P, Garcia-Lor A, Terol J, Curk F, Ollitrault F, Talón M et al (2015) Comparative values of SSRs, SNPs and InDels for citrus genetic diversity analysis. Acta Hort 1065:457–466
Ostrowski M-F, David J, Santoni S, Mckhann H, Reboud X, Le Corre V et al (2006) Evidence for a large-scale population structure among accessions of Arabidopsis thaliana: possible causes and consequences for the distribution of linkage disequilibrium. Mol Ecol 15:1507–1517
Otyama PI, Wilkey A, Kulkarni R, Assefa T, Chu Y, Clevenger J et al (2019) Evaluation of linkage disequilibrium, population structure, and genetic diversity in the U.S. peanut mini core collection. BMC Genomics 20:481–481
Pál C, Papp B, Hurst LD (2001) Does the recombination rate affect the efficiency of purifying selection? The yeast genome provides a partial answer. Mol Biol Evol 18:2323–2326
Park L (2019) Population-specific long-range linkage disequilibrium in the human genome and its influence on identifying common disease variants. Sci Rep 9:11380
Pavan S, Delvento C, Ricciardi L, Lotti C, Ciani E, D’Agostino N (2020) Recommendations for choosing the genotyping method and best practices for quality control in crop genome-wide association studies. Frontiers in Genetics 11
Phillips MA, Rutledge GA, Kezos JN, Greenspan ZS, Talbott A, Matty S et al (2018) Effects of evolutionary history on genome wide and phenotypic convergence in Drosophila populations. BMC Genomics 19:1–17
Plomion C, Bastien C, Bogeat-Triboulot M-B, Bouffier L, Déjardin A, Duplessis S et al (2016) Forest tree genomics: 10 achievements from the past 10 years and future prospects. Ann for Sci 73:77–103
Potts BM, Vaillancourt RE, Jordan G, Dutkowski G, Costa e Silva J, McKinnon G et al (2004) Exploration of the Eucalyptus globulus gene pool. In: Borralho N, Pereira J, Marques C, Coutinho J, Madeira M, Tomé M (eds) Eucalyptus in a changing world. Proceedings of an IUFRO conference, Aveiro, Portugal. RAIZ, Instituto Investigação de Floresta e Papel, pp 46–61
Pritchard JK, Przeworski M (2001) Linkage disequilibrium in humans: models and data. Am J Hum Genet 69:1–14
Quezada M, Aguilar I, Balmelli G (2022) Genomic breeding values’ prediction including populational selfing rate in an open-pollinated Eucalyptus globulus breeding population. Tree Genet Genomes 18:10
R Core Team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197:573–589
Ribeiro T, Barrela RM, Bergès H, Marques C, Loureiro J, Morais-Cecílio L et al (2016) Advancing Eucalyptus genomics: cytogenomics reveals conservation of Eucalyptus genomes. Frontiers in Plant Science 7
Schliep KP (2011) phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593
Shen C, Li X, Zhang R, Lin Z (2017) Genome-wide recombination rate variation in a recombination map of cotton. PLoS ONE 12:e0188682
Silva-Junior OB, Grattapaglia D (2015) Genome-wide patterns of recombination, linkage disequilibrium and nucleotide diversity from pooled resequencing and single nucleotide polymorphism genotyping unlock the evolutionary history of Eucalyptus grandis. New Phytol 208:830–845
Skelly DA, Magwene PM, Stone EA (2015) Sporadic, global linkage disequilibrium between unlinked segregating sites. Genetics 202:427–437
Slatkin M (2008) Linkage disequilibrium - understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–485
Slavov GT, DiFazio SP, Martin J, Schackwitz W, Muchero W, Rodgers-Melnick E et al (2012) Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol 196:713–725
Smith AV, Thomas DJ, Munro HM, Abecasis GR (2005) Sequence features in regions of weak and strong linkage disequilibrium. Genome Res 15:1519–1534
Soltis PS, Soltis DE (2021) Plant genomes: markers of evolutionary history and drivers of evolutionary change. PLANTS PEOPLE PLANET 3:74–82
Sperisen C, Büchler U, Gugerli F, Mátyás G, Geburek T, Vendramin G (2001) Tandem repeats in plant mitochondrial genomes: application to the analysis of population differentiation in the conifer Norway spruce. Mol Ecol 10:257–263
Steane DA, Conod N, Jones RC, Vaillancourt RE, Potts BM (2006) A comparative analysis of population structure of a forest tree, Eucalyptus globulus (Myrtaceae), using microsatellite markers and quantitative traits. Tree Genet Genomes 2:30–38
Szkiba D, Kapun M, Av H, Gallach M (2014) SNP2GO: functional analysis of genome-wide association studies. Genetics 197:285–289
Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME et al (2007) Recent human effective population size estimated from linkage disequilibrium. Genome Res 17:520–526
Thavamanikumar S, McManus LJ, Tibbits JFG, Bossinger G (2011) The significance of single nucleotide polymorphisms (SNPs) in Eucalyptus globulus breeding programs. Aust for 74:23–29
Thavamanikumar S, McManus LJ, Ades PK, Bossinger G, Stackpole DJ, Kerr R et al (2014) Association mapping for wood quality and growth traits in Eucalyptus globulus ssp. globulus Labill identifies nine stable marker-trait associations for seven traits. Tree Genet Genomes 10:1661–1678
Tibbits JFG, McManus LJ, Spokevicius AV, Bossinger G (2006) A rapid method for tissue collection and high-throughput isolation of genomic DNA from mature trees. Plant Mol Biol Report 24:81–91
Todesco M, Owens GL, Bercovich N, Légaré J-S, Soudi S, Burge DO et al (2020) Massive haplotypes underlie ecotypic differentiation in sunflowers. Nature 584:602–607
Tomé M, Almeida MH, Barreiro S, Branco MR, Deus E, Pinto G et al (2021) Opportunities and challenges of Eucalyptus plantations in Europe: the Iberian Peninsula experience. European Journal of Forest Research:1–22
Trickett AJ, Butlin RK (1994) Recombination suppressors and the evolution of new species. Heredity 73:339–345
Valenzuela CE, Ballesta P, Ahmar S, Fiaz S, Heidari P, Maldonado C et al (2021) Haplotype-and SNP-based GWAS for growth and wood quality traits in Eucalyptus cladocalyx trees under arid conditions. Plants 10:148
Vallejo RL, Silva RMO, Evenhuis JP, Gao G, Liu S, Parsons JE et al (2018) Accurate genomic predictions for BCWD resistance in rainbow trout are achieved using low-density SNP panels: evidence that long-range LD is a major contributing factor. J Anim Breed Genet 135:263–274
Wallis IR, Keszei A, Henery ML, Moran GF, Forrester R, Maintz J et al (2011) A chemical perspective on the evolution of variation in Eucalyptus globulus. Perspect Plant Ecol Evol Syst 13:305–318
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution:1358–1370
Wickham H (2016) ggplot2: Elegant graphics for data analysis. Springer-Verlag, New York
Wright S (1943) Isolation by distance. Genetics 28:114–138
Xing Y, Frei U, Schejbel B, Asp T, Lübberstedt T (2007) Nucleotide diversity and linkage disequilibrium in 11 expressed resistance candidate genes in Lolium perenne. BMC Plant Biol 7:43
Yeoh SH, Bell JC, Foley WJ, Wallis IR, Moran GF (2012) Estimating population boundaries using regional and local-scale spatial genetic structure: an example in Eucalyptus globulus. Tree Genet Genomes 8:695–708
Zhang W, Collins A, Gibson J, Tapper WJ, Hunt S, Deloukas P et al (2004) Impact of population structure, effective bottleneck time, and allele frequency on linkage disequilibrium maps. Proc Natl Acad Sci 101:18075–18080
Zimmerman SJ, Aldridge CL, Oyler-McCance SJ (2020) An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genomics 21:1–16
Acknowledgements
The authors wish to thank Forico Pty Ltd and Tree Breeding Australia (TBA) for access to the E. globulus trial and associated data. The authors thank the Chilean company Forestal Mininco SA (a subsidiary of Empresas CMPC SA) for permission to utilise the DNA of X46. The authors acknowledge the use of the high-performance computing facilities provided by Digital Research Services, IT Services at the University of Tasmania. The authors also wish to thank the anonymous reviewers for their comments on this manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This work was supported by the Australian Research Council (grant number DP190102053).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Communicated by: C. Rellstab
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Butler, J.B., Freeman, J.S., Potts, B.M. et al. Patterns of genomic diversity and linkage disequilibrium across the disjunct range of the Australian forest tree Eucalyptus globulus . Tree Genetics & Genomes 18, 28 (2022). https://doi.org/10.1007/s11295-022-01558-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-022-01558-7