
Abstract
Forest trees exhibit strong patterns of local adaptation in phenological traits along latitudinal gradients. Previous studies in spruce have shown that variation at genes from the photoperiodic pathway and the circadian clock are associated to these clines but it has been difficult to find solid evidence of selection for some of these genes. Here, we used growth cessation, gene expression, and single nucleotide polymorphism (SNP) data at two major candidate loci, FLOWERING LOCUS T/TERMINAL FLOWER1-Like2 (FTL2) and GIGANTEA (GI), as well as at background loci from a latitudinal gradient in Siberian spruce (Picea obovata) populations along the Ob River to test for clinal variation in growth cessation and at the two candidate genes. As in previous studies, there was a strong latitudinal cline in growth cessation that was accompanied by a significant cline in the expression of FTL2. Expression of FTL2 was significantly associated with allele frequencies at some of the GI’s SNPs. However, the cline in allele frequency at candidate genes was not as steep as in a Norway spruce cline and in a parallel Siberian spruce cline studied previously and nonsignificant when a correction for population structure was applied. A McDonald-Kreitman test did not detect decisive evidence of selection on GI (p value = 0.07) and could not be applied to FTL2 because of limited polymorphism. Nonetheless, polymorphisms contributed more to the increased neutrality index of PoGI than to that of control loci. Finally, comparing the results of two previously published studies to our new dataset led to the identification of strong candidate SNPs for local adaptation in FTL2 promoter and GI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A large number of plant and animal populations are locally adapted (Leimu and Fischer 2008; Savolainen et al. 2013). Local adaptation is usually shaped by multiple environmental factors, such as climate, photoperiod, soil conditions, or presence of parasites (Blanquart et al. 2013; Grøndahl and Ehlers 2008; Macel et al. 2007). If one factor dominates and is displayed along a geographical gradient, a cline in the adaptive trait may follow. Evidence of local adaptation in plants is common in herbaceous species that have relatively short generation time and experience strong selective pressures (Ågren and Schemske 2012; Hancock et al. 2011; Jain and Bradshaw 1966; Jakobsson and Dinnetz 2005; Joshi et al. 2001; Leimu and Fischer 2008). Compared to herbaceous plants, forest trees usually have substantially longer generation times, have large and continuously distributed population sizes, are wind pollinated, and experience extensive gene flow. All these biological and distribution characteristics suggest that it should, in principle, be more difficult to identify local adaptation in forest trees (Chambel et al. 2007; González-Martínez et al. 2002). Yet, local adaptation for phenological traits is actually very pronounced in many forest tree species (Lascoux et al. 2016; Savolainen et al. 2007). The contrast between strong local adaptation and weak genetic differentiation at quantitative traits despite important gene flow could, at least in part, be explained by the quantitative genetics model initially proposed by Le Corre and Kremer (2003) and recently extended by Berg and Coop (2014). Briefly, under this model, concerted small changes in allele frequencies at a large number of quantitative trait loci (QTL) rather than large allele frequency changes at a few of them can lead to important phenotypic differentiation even in the presence of gene flow. What this genetic architecture implies is that finding signatures of selection with classical tests will a priori be difficult.
One approach to enhance the power to detect local adaptation is to focus on variation along one or many environmental clines. Recent studies in forest trees have exhibited clinal variation at both phenotypic and genotypic levels across different geographic scales (Alberto et al. 2013; Brousseau et al. 2016; Chen et al. 2012; Oddou-Muratorio and Davi 2014; Pais et al. 2017; Savolainen et al. 2007). In studies of clinal variation and, more generally, in genome-wide association studies (GWASs), the analysis can be complicated by the presence of population structure/history which, if not properly accounted for, can lead to many false positives (Pritchard et al. 2010; Savolainen et al. 2011). On the other hand, if population structure co-varies with the cline, one can also face the opposite problem: namely, correcting for population structure will remove polymorphisms that are truly associated to the trait of interest, i.e., false negatives (Vilhjalmsson and Nordborg 2013). For instance, the postglacial re-colonization of conifer trees in Scandinavia through both a southern and a northeastern route generated a population genetic structure that could be very similar to the genetic differentiation created by adaptive selection along latitude (Chen et al. 2012; Pyhäjärvi et al. 2007). Population structure can be corrected, for instance, by using Bayesian generalized linear mixed models, but this may still lead to false negatives. In such circumstances, it is therefore important to replicate studies and use parallel clines in the same species, assuming that different parts of the natural range of the species had different and independent demographic histories (at least during the time period under consideration), or in different species, assuming that the same pathways underlie adaptation, to confirm the signatures of local adaptation (Chen et al. 2014; Yeaman et al. 2016).
During the Pleistocene and Holocene, Western Siberia was never completely glaciated, and forest trees persisted in Western and in Central Siberia. During the Late Pleistocene, Western Siberia was mostly a cold desert with relicts of arboreal vegetation in large river valleys (Velichko et al. 2011). Indeed, paleoecological data indicate that spruce and other tree species survived in local refuges such as sand dunes, mountains, and valleys in Western Siberia from prior to the Last Glacial Maximum (LGM), through the LGM and Late Glacial and to the Holocene (Binney et al. 2009; Väliranta et al. 2011). These relict vegetated areas, including forested ones, were likely rather large as extensive fossils of large animals were also found in these regions (Kosintsev et al. 2012). A likely consequence of the history of the region is that population genetic structure can be very weak over very large regions. For example, population genetic structure along a cline following the Yenissei River and running over 10° of latitude is barely detectable (Chen et al. 2014), even with hundreds of thousands of SNPs (C. Chen, M. Lascoux, and P. Milesi, unpublished data). These latitudinal clines are, therefore, extremely useful if one aims to detect signature of local adaptation for traits related to latitude such as phenological traits in plants that are controlled by photoperiod (Lotterhos and Whitlock 2015).
Since the seminal study of Dormling (1973) and Savolainen et al. (2011), a large body of work has shown that growth cessation and budset are controlled by photoperiod. More recent studies have started to identify some of the genes underlying the variation in growth cessation. In both populations of Norway spruce (Picea abies) from Scandinavia and Siberian spruce (Picea obovata) from the Yenisei River, a clear clinal pattern in allele frequency and/or expression level was observed at two candidate genes: FLOWERING LOCUS T/TERMINAL FLOWER1-Like2 (FTL2) and GIGANTEA (GI) (Chen et al. 2012, 2014). The FTL2 gene is associated with growth cessation, its level of expression strongly correlates with latitude, and when overexpressed in P. abies, it led to budset (Chen et al. 2012, 2014; Gyllenstrand et al. 2007; Karlgren et al. 2013). FT-Like genes integrate signals from the different pathways controlling flowering time in plants (Fornara et al. 2010; Pin et al. 2010; Pin and Nilsson 2012), and studies have shown that they also play a major role in the control of phenology in trees (Avia et al. 2014; Opseth et al. 2016; Wang et al. 2018). Changes in FTL2 expression around procambium and vascular tissues and that in the crown region in buds in Norway spruce were shown to be significantly associated with growth cessation, which differs drastically between northern and southern populations (Gyllenstrand et al. 2007; Karlgren et al. 2013). Chen et al. (2012, 2014) suggested that a SNP in the promoter of PaFTL2 might affect the divergent expression patterns observed between genotypes. Previous studies in Arabidopsis thaliana indicated that FT homologs control growth cessation through cis-regulatory changes (Schwartz et al. 2009; Adrian et al. 2010) and that the length of the FT promoter varies widely through insertions and deletions to adapt to certain light length and temperature (Liu et al. 2014). GI, a plant-specific nuclear protein, plays a major role in the photoperiodic pathway and the regulation of circadian clock and also affects many physiological processes in plants (de Montaigu et al. 2015; Ding et al. 2018; Mishra and Panigrahi 2015; Zhou et al. 2018). GI is presumably located upstream of the FTL2 gene (Holliday et al. 2010). In A. thaliana, daily rhythms of GI expression responded to day length and its sensitivity to day length is significantly correlated with latitude. It was shown that the latitudinal cline in GI expression resulted from an increased delay in response to longer spring photoperiods in southern accessions (de Montaigu and Coupland 2017). In previous studies of P. abies and P. obovata, GI exhibited strong clinal variation of allele frequency but weak expression differentiation. SNPs within GI have also shown significant signals of diversifying selection based on FST test; however, direct evidence of selection was limited since classical tests could not be carried out due to a very limited number of polymorphisms, in particular synonymous changes (Chen et al. 2012, 2014). The latter could be the consequence of recurrent episodes of selective sweeps as identified in poplar (Hall et al. 2011; Keller et al. 2012). Finally, although one suspects that GI acts upstream of FTL2, the exact nature of the relationship between the two genes is not known in spruce.
The aims of the present study were twofold. First, we wanted to test whether the clinal variation in growth cessation and in FTL2 expression that was previously observed in Norway spruce (along a Scandinavian cline, Chen et al. 2012) and Siberian spruce (along a Yenisei River cline, Chen et al. 2014) was also observed along an Ob River cline. Our second aim was to test for clinal variation in allele frequency and for the presence of selection at the GI and FTL2 loci. To increase our chance to reach these goals, the current study differs from previous ones in two essential ways. Firstly, the cline along the Yenisei River studied in Chen et al. (2014) had a low coverage of high-latitude zones (those above 60° N). However, it is above this latitude that one observes the steepest change in growth cessation (Chen et al. 2014). Samples from populations at latitudes exceeding 60° N would therefore provide more power to detect changes in allele frequencies at FTL2 and GI, if those are indeed related to growth cessation. In the present study, we collected samples along the Ob River in Western Siberia between 58° N and 67° N to test for the presence of selection on FTL2 and GI. Secondly, as noted above, the fragments of GI that were sequenced in previous studies were short and had very few synonymous and nonsynonymous sites, leading to problems in the implementation of neutrality tests to the gene. To circumvent this problem, we sequenced GI fragments four times as long as in previous studies, in order to gather more synonymous and nonsynonymous polymorphisms and increase our power to detect signature of selection.
Materials and methods
Sample collection and growth cessation measurement
We collected seeds of Siberian spruce (P. obovata) from individual trees in seven populations along the Ob River, from latitude 58° N to latitude 67° N (Fig. 1, Table 1). Healthy seeds from different mother trees in each population were germinated in Petri dishes after being soaked in water at 4 °C for 48 h. Seedlings at 20-needle stage were transplanted in plastic pots filled with a mixture of expanded clay aggregate (LECA, Sweden) and humus (1:3 in volumes). Four seedlings from different maternal trees, but of the same population, were randomly planted in each pot. On average, 6–72 seedlings from 3 to 15 maternal trees were planted for each population in a growth chamber with a temperature of 18 °C, 54% humidity, and continuous light with a PAR value of 84 μE for 8 weeks. Thereafter, seedlings were exposed to photoperiodic treatments of increasing night length. Each photoperiodic length period lasted for 1 week starting with a first week with continuous (24 h) light. A night length of 2 h was introduced in the second week and was then extended by 1.5 h every week until the photoperiod reached 14.5-h light/9.5-h dark. The final treatment lasted for 2 weeks, and the whole photoperiodic treatments lasted 8 weeks (Chen et al. 2014).

Population distribution in three regions referred to in the article: Ob River, Yenisei River (Chen et al. 2014), and Scandinavia (Chen et al. 2012). Colors represent different clines. The distribution shape of Picea abies was downloaded from the EUFORGEN website (www.euforgen.org), and the distribution of Picea obovata was based on Lockwood et al. (2013)
Seedling height was measured twice a week before the start of the photoperiodic treatment. After the treatment started, measurements were taken once a week at the end of each photoperiodic period (see Chen et al. 2014). We used growth cessation as a proxy for budset. Growth cessation was defined as the date on which the weekly height increase was less than 5% of the total plant height to account for measurement error. We counted the growing days until the plant stopped growing after the photoperiodic treatment started. The “relative number of growing days” was defined as the number of growing days divided by the total number of days of the photoperiodic experiment and used for linear regression on population latitudes. The slopes of the linear regression were compared between the Ob River and the Yenisei River clines (data collected from Chen et al. 2014) to test whether there were significant differences in the associations between the two regions using the ANOVA function in R 3.3.1 (R Core Team 2016).
PoFTL2 expression
The expression level of the FTL2 gene correlated with differences in growth cessation between southern and northern populations of P. abies (Gyllenstrand et al. 2007). A mutation within its promoter region exhibited the strongest latitudinal gradient in frequency among candidate genes and was shown to be under adaptive selection in both P. abies and P. obovata (Chen et al. 2012, 2014). In this study, we measured the expression level of PoFTL2 gene for the Ob River populations in order to validate the variation observed along the Yenisei River in an independent cline.
Because the expression peak of PoFTL2 fluctuated under daylight (Chen et al. 2012), needles were sampled twice at 9 am and 5 pm during the last 24 h of each photoperiodic treatment and the expression values at these two time points were averaged to reduce sampling error. Samples at each time point were composed of two replicates, with the exception of populations Berezovo (BER-64) and Pytier (PUT-66), where no replicates were collected because of low germination rates. For the two northernmost populations Kamys Mys (KAZ-65) and Krasnij Kamenj (KK-67), we only succeeded in sampling once at 9 am and without replicates because of early growth cessation (Table S1). Four needles were pooled together from different seedlings in each population for each replicate at each time point. Each time needles were collected from different individuals but in the same population in order to minimize the health impact on seedlings. Total RNA was extracted separately from each sample following the instruction of STRN250 Spectrum Plant Total RNA Kit (Sigma-Aldrich, Saint Louis, USA) and quantified before PCR. Complementary DNAs (cDNAs) were synthesized from 0.5 μg total RNA using Superscript III reverse transcriptase (Thermo Fisher, Waltham, MA, USA) and random hexamer primers. For each sample, the reaction was conducted in duplicates with UBQ gene as a control gene. Prepared cDNA was diluted to 1:100 in volume and mixed with a reaction mixture of 5 μl DyNAmo Flash SYBR Green (DyNAmo Flash SYBR Green qPCR Kit; Finnzymes, Espoo, Finland) and 0.5 μl each FTL2/UBQ primer (see Chen et al. 2012 and Gyllenstrand et al. 2007 for a detailed protocol and primer sequences). RT-qPCR was carried on an Eco Real-Time PCR instrument (Eco™ software; Illumina, San Diego, CA, USA) with thermal parameters: polymerase activation at 95 °C for 7 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s, and melt curve lasted for 15 s at 95°, 55°, and 95°, respectively. The expression levels of population x at each time point t after photoperiodic treatment i were calculated as ΔCTxi (t) = CTreference xi (t) − CTtarget xi (t), where CTreference means the average Cq value of the UBQ gene, and CTtarget refers to that of PoFTL2 gene. For each population, we took the average ΔCT values of two replicates under each treatment. To compare expression levels, the relative expression level is defined as R(xi)(t) = (ΔCTxi(t) − ΔCTSN), where ΔCTSN is the average ΔCT value under 24-h-light treatment of the southernmost population (TOB) (see Chen et al. 2012 for additional details). Estimates of the expression level of PoFTL2 for all Ob River populations were regressed on latitude using the lm function in R 3.3.1 (R Core Team 2016). Then, we used ANOVA to compare the PoFTL2 expression under each treatment between the Ob River cline and the published Yenisei River cline (Chen et al. 2014).
DNA extraction and sequence preparation
DNA was extracted from 87 megagametophytes (Table 1) that only contain haploid maternal DNA, using the DNeasy Plant Mini Kit (Qiagen, Germantown, MD, USA). Two candidate genes, together with 14 control fragments, were amplified using Phusion DNA polymerase and sequenced using Sanger sequencing technology. The two candidate genes were PoFTL2 and PoGI. PoFTL2 included part of the promoter region, and PoGI contained 8 fragments (PoGI5723, PoGI6705, PoGI14199, PoGI23280, PoGI25676, PoGI28689, PoGI41355, and PoGI44900; Table S2). The fourteen control loci (Can8a, Can12, Can14, Can28, Can31, Can32, Can33, Can37, Can49, Can56, Can58, Can59, Can60, and Can62) were randomly chosen from genes of unrelated functions (Pavy et al. 2012). BLAST search against GenBank and UniProt databases suggested that these loci are unrelated to phenology and fitness and they were included here to correct for demographic effects. We used the Phred/Phrap/Consed programs (Ewing and Green 1998; Ewing et al. 1998; Gordon et al. 1998) for base calling, contig assembly, and sequence editing. Base quality was manually examined, and sites with a quality score lower than 20 were considered as missing. Sites were also excluded from further analyses if more than two alleles were identified.
Genetic diversity, linkage disequilibrium, and population structure
For each gene, we estimated the number of segregating sites (S), the pairwise nucleotide diversity (π) (Nei and Li 1979), Watterson’s estimate of the scaled population mutation rate (θw) (Watterson 1975), and Tajima’s D (Tajima 1989) for both coding and noncoding regions. Linkage disequilibrium (LD) between all pairs of SNPs within a gene was calculated for the whole dataset. SNPs of the same gene were considered as significantly linked and grouped together when r2 ≥ 0.25 and Bonferroni-corrected p value ≤ 0.05. The overall decay of LD with physical distance within genes was calculated by nonlinear regression of r2 on a distance between polymorphic sites measured in base pairs, using the formula suggested by Remington et al. (2001). We then used 216 unlinked silent SNPs from all loci to assess population genetic structure using STRUCTURE V2.3.4 analysis (Hubisz et al. 2009; Pritchard et al. 2000). Ancestral components of individual genotypes were estimated by varying the number of clusters in the dataset (K) from 1 to 7, using an admixture model with correlated allele frequencies. Ten independent runs were conducted for each value of K, and each run was composed of a burn-in of 100,000 iterations and additional 1,000,000 iterations. The ΔK criterion (Evanno et al. 2005) and the log posterior probabilities LnPr(X|K) (Pritchard et al. 2000) were used to determine the most likely value of K. According to Janes et al. (2017), population genetic structure may be underestimated for highly admixed populations and, in general, estimating K is fraught with difficulties. In order to gain trust in our results, we compared the results to those in Chen et al. (2014) who analyzed a similar cline in the same species along the Yenisei River.
Analysis of clinal variation in allele frequencies
Different methods, including linear regression and Bayenv2, were used to test the association between allele frequencies and latitude and are described below. In general, for each tested summary statistics, control SNPs from the background loci were used to build an empirical distribution. Then, we selected outlier SNPs at the 10% and 5% tails of this empirical distribution and tested for enrichment of SNPs in candidate genes (PoGI and PoFTL2) by comparing the ratios of candidate over control SNPs at different significance levels to the ratio of the original dataset, which is equal to 0.76.
Linear regression on latitude
Allele frequencies were calculated for each population and were transformed using a square root of arcsine function (Berry and Kreitman 1993). The transformed allele frequencies were then regressed on latitude using the “lm” function in R 3.3.1 (R Core Team 2016). We used the coefficient of determination, the adjusted r2, as a statistic for “clinality” to measure the proportion of the total variance of allele frequency that could be explained by latitude (Berry and Kreitman 1993).
Bayesian generalized linear mixed-model analysis
To correct for the effect of population structure when assessing the correlation between allele frequency and latitude, we used a Bayesian generalized linear mixed model implemented in the program Bayenv2 (Coop et al. 2010; Günther and Coop 2013). In the null model, allele frequency of each population follows a multivariate normal distribution that deviates around the mean value (a common ancestor) and correlates by a variance-covariance matrix, which accounts for population structure generated by random genetic drift (Nicholson et al. 2002). The alternative model incorporates an environmental or geographic effect as a fixed linear effect (Coop et al. 2010; Hancock et al. 2011). To test for the support of the environmental factor, the program uses a Bayes factor (BF) that compares the posterior probabilities for the alternative and null models. Two hundred two unlinked control SNPs were used to estimate the covariance matrix whereas BFs were computed for all SNPs. In order to make sure the covariance matrix converged, we compared the estimates of six independent runs. BF results were averaged across six runs of 1,000,000 iterations.
Association between PoFTL2 expression and allele frequencies
To test the association between PoFTL2 expression and allele frequencies, we used the same model as the one implemented in Bayenv2 (Coop et al. 2010; Günther and Coop 2013). Values of PoFTL2 expression in each light period and population were standardized and treated as the dependent variable. As mentioned above, to avoid false positives caused by population structure, the mean covariance matrix of allele frequencies across unlinked control silent loci was estimated through six Monte Carlo Markov chains and each with 1,000,000 iterations. For each SNP, six MCMCs of 1,000,000 iterations were run to estimate the BF.
Selection tests
To examine whether the observed latitudinal variation was caused by diversifying selection, we first applied an FST outlier test using the program BayeScan v. 2.1 (Foll and Gaggiotti 2008; Foll et al. 2010; Fischer et al. 2011). This Bayesian approach is based on an island model to estimate the variance of gene flow between subpopulations, which seems a reasonable assumption in our case based on the STRUCTURE results (see “Results” for details). In the model, FST has two components: a population-specific component shared by all loci and a locus-specific component shared by all populations. The alternative model for a given locus is retained when the locus-specific component significantly differs from zero, a positive value of which suggests diversifying selection if a large FST value is observed as well. We ran BayeScan with 20 pilot runs and a burn-in of 50,000 steps followed by 50,000 output iterations. The prior odds ratio was set to 1, which is approximately the ratio of candidate/control SNPs. We also tested a prior odds ratio equal to 10, which is more stringent to reduce possible false positives. FST outliers were selected at the 10% and 5% tail of log10 (q value) adjusted for multiple comparisons.
Finally, we also applied a McDonald-Kreitman neutrality test (McDonald and Kreitman 1991) to PoGI (one could not do it for PoFTL2 as the number of polymorphic sites was too limited). We first estimated the number of sites that were either polymorphic (P) or divergent (D) at nonsynonymous sites (n) and silent sites (s, including synonymous and noncoding sites). Sequences of Picea breweriana were used to estimate divergence. The neutrality index (Rand and Kann 1996; Stoletzki and Eyre-Walker 2011) is defined as (Pn/Ps)/(Dn/Ds): a value of NI greater than 1 indicates the presence of slightly deleterious mutations or balancing selection and a value less than 1 indicates positive selection (Cutter 2019, p. 186). We also compared the NI values of control loci to the value obtained for PoGI. To reduce the statistical error caused by rather short fragments for each control locus (~ 520 bp), we calculated an averaged NI value across control loci by concatenating all control loci and resampling the same number of nucleotides as in PoGI. In total, 7300 bp was sequenced in control loci and 12,000 bp were resampled with replacement when compared to PoGI.
Results
Clinal variation of growth cessation
The number of relative growing days declined significantly as latitude increased (adjust r2 = 0. 8151, p value = 0.003). Trees from the southernmost population ceased growing much later than those of the northernmost one, and the ratio of the relative number of growing days of the southernmost population to that of the northernmost one was around 9 (Fig. 2). For comparison, we also included growth cessation data from the Yenisei River (from 54° N to 66° N; Fig. 1; Chen et al. 2014) and performed a combined analysis. The linear regressions showed a slightly steeper decline trend in the number of growing days with latitude in the Ob River dataset (reg. coef. = − 0.031, p value = 0.003) than in the Yenisei River (reg. coef. = − 0.022, p value = 0.001), but the difference was not significant (p value = 0.11 for the interaction effect between both datasets).

Relative number of growing days for Siberian spruce populations along the Yenisei River (Chen et al. 2014) and Ob River, estimated in growth chamber experiments. Populations along the Ob River are represented by blue dots, and populations along the Yenisei River are by red ones. Vertical bars represent the standard deviation for each measurement. The dark gray regions show the 95% confidence interval for the relative growing days of each population
PoFTL2 gene expression
The expression of PoFTL2 increased as night length was extended in all Ob River populations (Fig. 3a). PoFTL2 expression was significantly correlated to latitude under treatments at 19-h light, 17.5-h light, 16-h light, and 14.5-h light (Table S3). We used an ANOVA to compare PoFTL2 expression under each treatment in the Ob River populations and already available expression data from the Yenisei River (Chen et al. 2014) (Fig. 3b). For all photoperiodic treatments, the coefficients did not differ significantly between the two clines (Table S3).

PoFTL2 relative expression pattern. left panel Seedlings from the Ob River populations and right panel seedlings from the Yenisei River populations during the growth cessation experiment (Chen et al. 2014). Colors represent eight successive light treatments
Genetic diversity, linkage disequilibrium, and population genetic structure
We sequenced a total length of 12,000 bp for PoGI, containing 1310 bp coding sites, which covered 24% of the GI sequence in the current P. abies reference genome (Nystedt et al. 2013) and 42% of the whole coding regions, 2800 bp for PoFTL2 including a 1151-bp promoter region and a 198-bp coding region, and 7300 bp for all 14 control loci carrying 3866-bp coding sites; 62 to 87 individuals were successfully sequenced and aligned. In total, 371 SNPs were extracted from all loci, including 104 for PoGI, 56 for PoFTL2, and 211 for the 14 background genes. PoGI and PoFTL2 had similar π and θw values in noncoding regions (PoGI: π = 0.0012 and θw = 0.0020; PoFTL2: π = 0.0018 and θw = 0.0013), which were both lower than the averages over the control loci (average π = 0.0058; θw = 0.0065). However, the PoFTL2 promoter had a higher π value (average π = 0.0084). For coding regions, both π and θw values were lower in PoGI than in control loci and no polymorphisms were detected in the PoFTL2 coding region. Tajima’s D estimates were close to zero for both control and candidate genes (Table 2).
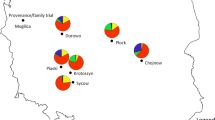
LD was estimated using r2 decay within genes. In general, r2 decreased below 0.1 within 250 bp (Fig. 4). The median and mean value of r2 was equal to 0.009 and 0.1, respectively. There were 37 significantly linked LD groups for control SNPs and 19 for candidate SNPs (Bonferroni-corrected p value ≤ 0.05 and r2 ≥ 0.25). As in the analysis of the Yenisei River cline by Chen et al. (2014) the STRUCTURE results showed that nearly all individuals were genetically admixed and little difference could be identified between populations (Fig. 5), based on the optimal number of clusters which was K = 3 which only added additional admixture compared to K = 2, but did not improve clustering (Fig. S1).

Pairwise estimates of the linkage disequilibrium (LD) decay between loci in Picea obovata. The red curve shows the mean decay of r2 with distance

Results of the population clustering analysis based on unlinked silent SNPs. The population genetic structure plot for K = 2 and K = 3 in Picea obovata is shown
Clinal variation analyses
Twenty SNPs (16 from PoGI and 4 from PoFTL2) showed significant clinal variation in allele frequency along latitude (adjusted r2; p ≤ 0.05). In the enrichment test of candidate SNPs, 26 and 20, candidate outliers were found at the 10% and 5% tails of the empirical distribution of adjusted r2, respectively. The corresponding ratios of candidate to control outliers were 2.37 and 2. Compared to the original ratio of candidate to control SNPs (0.76), enrichment of candidate SNPs was found at both 10% and 5% tails (Fisher’s exact test, p ≤ 0.05) (Table 3).
We corrected for the possible effect of population structure on clinal variation analyses by introducing a covariance matrix in the Bayesian generalized linear mixed model that tests environmental effects on allele frequency changes (Bayenv2). None of the variation at candidate loci and control loci was significantly correlated with latitude when a threshold of BF > 3 was used.
F ST outliers
We applied BayeScan to search for SNPs under divergent selection. The median of locus-specific FST values was 0.0397. There were no significant outliers once a FDR correction was applied.
Neutrality test on PoGI and PoFTL2
Finally, we conducted a McDonald-Kreitman test on PoGI. No MK test could be carried on PoFTL2 as polymorphism was too limited. For PoGI, the NI value was 2.33 and the corresponding Fisher’s exact test p value was 0.078 (Table 4). For the concatenated control loci, the NI value was 2.03. We then compared polymorphism ratios and divergence ratios between PoGI and control loci and found that the polymorphism ratio contributed more than the divergence ratio to the increased NI value in PoGI.
Association between FTL2 expression and polymorphisms
After correction for population structure, allele frequencies at 4 control SNPs and 6 SNPs in PoGI were significantly associated with PoFTL2 expression. In the case of the SNP from PoGI, half of these associations occurred during treatment 6, i.e., at the start of the last photoperiodic treatment when the night length had reached 9.5 h/day (Table 5).
Comparing SNPs among clines
Evidence for clinal variation in allele frequency or presence of selection was much weaker along the Ob River cline than in our two previous studies (Chen et al. 2012, 2014). In this study, we found 33 SNPs in PoFTL2 and PoGI genes with marginal evidence for clinal variation or presence of selection in one or more analyses (Table S4). In order to investigate whether the same genes or even identical SNPs could indeed be good candidate loci for local adaptation and also to rule out the effect of demography in an easier way, we compared the results of the current study to two other clines covering a similar latitudinal range: the Yenisei River cline of P. obovata (Chen et al. 2014) and the Scandinavian cline of P. abies (Chen et al. 2012) which had quite different post-glacial history compared to the Ob River cline. For FTL2, six SNPs out of a joint total number of 119 from its promoter were shared among all three clines (Table S5). Two of them (FTL2 promoter_87 and FTL2 promoter_332) showed clinal variation with latitude in all three studies, with FTL2 promoter_87 exhibiting the strongest correlation with latitude along the Ob River cline (Table S4). Eight SNPs within the FTL2 promoter were shared exclusively between the Ob River and the Yenisei River clines, and five were shared between the Ob River and the Scandinavian clines. In the FTL2 coding region, only one SNP was shared by all three clines (FTL2_78), four were exclusively shared between the Ob River and the Yenisei River clines, and five were common between Ob River and the Scandinavian clines, respectively. No mutations were exclusively shared between the Yenisei River cline and the Scandinavian cline. For the GI fragment, four common SNPs were found in all clines, including two nonsynonymous substitutions that showed the strongest selection signal: GI5723_572 and GI23280_669. At GI5723_572, 24.5% of histidine (His) changed into tyrosine (Tyr) in the southern Siberian populations and 56.5% of the amino acids were Tyr in the northern Siberian populations. This mutation also exhibited the strongest selective signals in the two other clines [see GIF2_9_987 in Chen et al. 2012 and GIF2_605 in Chen et al. 2014] and possibly causes a difference in peptide folding based on protein structure prediction (Chen et al. 2014). Furthermore, eleven GI SNPs were exclusively shared between the Ob River and the Yenisei River clines, including two nonsynonymous changes.
Discussion
The present study is part of a series of studies on local adaptation in two closely related spruce species (Heuertz et al. 2006; Gyllenstrand et al. 2007; Chen et al. 2012, 2014; Karlgren et al. 2013). As other studies in forest trees [e.g., Hall et al. 2011 and Wang et al. 2018 in Populus tremula, Avia et al. 2014 in Pinus sylvestris, and Grivet et al. 2011 in Pinus pinaster and Pinus halepensis], it is worth recalling that this series of studies started as candidate gene studies, with candidate genes putatively associated to phenology in forest trees being selected among genes with a proven involvement in the control of flowering time in A. thaliana (Fornara et al. 2010). Perhaps surprisingly, given the 300 Mya separating gymnosperms and angiosperms (Doyle 2012), this candidate gene approach has been rather successful in conifers (see also Avia et al. 2014 for an example in Pinus sylvestris) demonstrating the association of candidate gene polymorphisms with, and functional involvement in, the variation in phenology. For some genes, it was even possible to show that these genes were under natural selection (Chen et al. 2014; Wang et al. 2018), but this has been more difficult for other genes, like GI. The present study provides further support for the usefulness of a candidate gene approach in nonmodel species, especially those with large and still poorly characterized genomes such as spruce. The aim of the present study was twofold. First, we tested whether the clinal variation in growth cessation and in gene expression in genes related to the control of growth cessation that was previously observed in Norway spruce (the Scandinavian cline) and Siberian spruce (the Yenisei River cline) (Chen et al. 2012, 2014) was also observed along an Ob River cline. The Ob River is located close or within the hybrid zone between P. abies and P. obovata (Tsuda et al. 2016), and the cline under study is situated at latitudes at which the steepest change in growth cessation was observed along the Yenisei River cline, where we unfortunately had only a few populations (Chen et al. 2014). Second, in our two previous studies (Chen et al. 2012, 2014), we could not properly test for the presence of selection on GI due to a lack of synonymous changes in the limited part of the gene that was studied. We therefore sequenced around 12,000 bp of the GI gene, which is four times longer than the 3000 bp sequenced in our previous studies (Chen et al. 2012, 2014). Our results confirm the presence of a cline in growth cessation and the existence of a latitudinal gradient in the expression pattern of FTL2 and suggest the presence of an association between polymorphism in GI and the expression of FTL2. The cline in allele frequencies at both GI and FTL2 was present but was weaker than that in previous studies and altogether nonsignificant when we corrected for population structure. Finally, selection may have affected polymorphism at GI, even if a clear pattern of selection at GI remains elusive. Below, we will discuss the limits and implications of these results.
Clinal variation in growth cessation, FTL2 gene expression, and allele frequencies at GI and FTL2
As in our previous studies (Chen et al. 2012, 2014), we found significant clinal variation in growth cessation and FTL2 expression along a latitudinal gradient. The present study, hence, strengthens the status of FTL2 as a major gene involved in the control of growth cessation in trees (Wang et al. 2018). It also links, for the first time, expression at FTL2 with polymorphism at GI, since allele frequencies at SNPs within GI were associated to the level of expression of FTL2 at the end of the photoperiodic treatment. How variation in GI influences downstream genes involved in growth-related traits or phenology is not yet very clear: while its expression seems ubiquitous suggesting a highly pleiotropic role (Mishra and Panigrahi 2015), in A. thaliana, natural variation in GI expression was correlated with growth traits but neither with CONSTANS expression nor with flowering (de Montaigu and Coupland 2017).
Evidence of clinal variation at SNPs in both GI and FTL2, though present, was much weaker than that in the case of the Scandinavian or Yenisei River clines and, generally, nonsignificant. Even for SNPs in GI and FTL2 that showed significant clines, the enrichment ratios of candidate to control SNPs in the Bayenv2 analysis were not significant. One possible cause is simply that the number of candidate and control genes as well as the number of individuals of the present study are more limited than those in Chen et al. (2012) and Chen et al. (2014) and that the cline is shorter, although since the cline along the Ob River resembles that along the Yenisei River adding more populations under 58° N should not have increased much the variation in growth cessation. Alternatively, the weaker clinal variation in allele frequency could be due to the fact that the present cline considers a set of populations located at higher latitude or that the Ob River is part of the hybrid zone between P. abies and P. obovata. The former, however, does not seem very likely as the cline in growth cessation is more pronounced in this study than in the study on the Yenisei River cline. Similarly, the latter is not supported by the fact that the cline in Scandinavia, which is much more recent, is also more pronounced. The level of linkage disequilibrium was also similar to the values observed in Siberian spruce populations along the Yenisei River (median r2 = 0.006), Norway spruce in Scandinavia (average r2 = 0.2), and white spruce (Beaulieu et al. 2011; Chen et al. 2012, 2014). This low level of LD might simply reflect the fact that the hybrid zone is a rather ancient one as Tsuda et al. (2016) estimated the split between P. obovata and the hybrid zone between 0.2 and 2 Mya. Another possible reason is that natural selection could act on different paths to establish local adaptation since growth cessation is a quantitative trait. In addition to GI and FTL2, other photoperiodic candidate genes were related to growth cessation and/or under natural selection such as, for instance, PHYP, PRR3, PRR7, and CCA1 (Chen et al. 2012; Källman et al. 2014). A larger role of these genes in the Ob River cline could weaken the effect of selection on GI and FTL2.
Selection on GI
GI is a key component of the circadian clock in plants, in general, and it plays a major role in the control of phenology in trees (Alberto et al. 2013; Holliday et al. 2010; Keller et al. 2012). It has, however, been difficult to characterize its genetic variation due to its length and low and peculiar polymorphism, with very limited synonymous changes and mostly nonsynonymous ones (Chen et al. 2012, 2014; Keller et al. 2012). In this study, we sequenced a much larger part of the gene and found suggestive evidence of selection on GI, with an excess of nonsynonymous polymorphism at GI compared to background loci and a higher neutrality index. Our study is the second one suggesting the presence of selection in GI, albeit weak, in forest trees. In Populus tremula, Hall et al. (2011) studied variation at two copies of GIGANTEA GIA and GIB and found some evidence of selection on both genes, with, as in our case, an excess of nonsynonymous polymorphisms. Based on patterns of polymorphisms and divergence, in particular a negative correlation between synonymous nucleotide diversity and estimated selection intensity on nonsynonymous changes, Hall et al. (2011) suggested that the pattern of diversity at photoperiodic genes could be explained by recurrent hitchhiking selective sweeps. Our data does not allow us to be more specific on the type of selection acting on GI, but recurrent hitchhiking selective sweeps are certainly a possible scenario. A larger study than the present one, sequencing the whole GI gene across a number of species and considering both polymorphism and divergence, would be needed to find more definitive evidence of selection on GI.
General implications: potential and inherent limits of clinal variation studies
The study of latitudinal clines has been one of the main sources of information on local adaptation in forest trees (Hall et al. 2011; Wang et al. 2018; Yeaman et al. 2016) and the use of parallel clines within the same species or in different species, a practical and elegant way to control for the effect of population structure. Strikingly, studies in both conifers and angiosperms have pointed to the same group of genes, so far mostly genes from the photoperiodic pathway, in particular FT-Like genes, suggesting that the same mechanism may be at work in groups of plants that have diverged hundreds of million years ago. In Norway spruce, the availability of thousands of polymorphisms across the genome of thousands of individuals from the Swedish breeding program, together with similar data from latitudinal and longitudinal clines, offers a unique opportunity to obtain a better understanding of the genetic basis of local adaptation to latitude and assess the potential of assisted migration to counter the effect of climate change (Aitken and Whitlock 2013; Milesi et al. 2019). On the other hand, as alluded to in the “Introduction,” there are strong inherent limits to the dissection of quantitative adaptive traits such as growth cessation. In a genome-wide association study based on 1500 spruce trees from Southern Sweden, we found that 32 SNPs belonging to 15 transcripts were associated to variation in budburst, an important phenology trait, which is primarily controlled by temperature (Milesi et al. 2019). While budburst was less polygenic than height or diameter (131 and 138 transcripts associated to trait variation, respectively), it still appears as a very polygenic trait. The study design did not allow the estimation of the effect of individual loci but likely those were limited. As exemplified by the findings of the numerous, large-scale studies that have attempted to dissect the genetic basis of flowering time in A. thaliana, it will be difficult to go beyond the characterization of a limited number of key loci (The 1001 Consortium 2016).
References
Adrian J, Farrona S, Reimer JJ, Albani MC, Coupland G, Turck F (2010) cis-Regulatory elements and chromatin state coordinately control temporal and spatial expression of FLOWERING LOCUS T in Arabidopsis. Plant Cell 22:1425–1440
Ågren J, Schemske DW (2012) Reciprocal transplants demonstrate strong adaptive differentiation of the model organism Arabidopsis thaliana in its native range. New Phytol 194:1112–1122
Aitken SN, Whitlock MC (2013) Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol Syst 44:367–388
Alberto FJ, Aitken SN, Alía R, González-Martínez SC, Hänninen H, Kremer A, Lefèvre F, Lenormand T, Yeaman S, Whetten R, Savolainen O (2013) Potential for evolutionary responses to climate change—evidence from tree populations. Glob Chang Biol 19:1645–1661
Avia K, Kärkkäinen K, Lagercrantz U, Savolainen O (2014) Association of FLOWERING LOCUS T/TERMINAL FLOWER 1-like gene FTL2 expression with growth rhythm in Scots pine (Pinus sylvestris). New Phytol 204:159–170
Beaulieu J et al (2011) Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics 188:197–214
Berg JJ, Coop G (2014) A population genetic signal of polygenic adaptation. PLoS Genet 10:e1004412
Berry A, Kreitman M (1993) Molecular analysis of an allozyme cline: alcohol dehydrogenase in Drosophila melanogaster on the east coast of North America. Genetics 134:869–893
Binney HA et al (2009) The distribution of late-Quaternary woody taxa in northern Eurasia: evidence from a new macrofossil database. Quat Sci Rev 28:2445–2464
Blanquart F, Kaltz O, Nuismer SL, Gandon S, Ebert D (2013) A practical guide to measuring local adaptation. Ecol Lett 16(9):1195–1205
Brousseau L et al (2016) Local adaptation in European firs assessed through extensive sampling across altitudinal gradients in Southern Europe. PLoS One 11:e0158216
Chambel MR, Climent J, Alía R (2007) Divergence among species and populations of Mediterranean pines in biomass allocation of seedlings grown under two watering regimes. Ann Forest Sci 64:87–97
Chen J et al (2012) Disentangling the roles of history and local selection in shaping clinal variation of allele frequencies and gene expression in Norway spruce (Picea abies). Genetics 191:865–881
Chen J et al (2014) Clinal variation at phenology-related genes in spruce: parallel evolution in FTL2 and Gigantea? Genetics 197:1025–1038
Coop G, Witonsky D, Di Rienzo A, Pritchard JK (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics 185:1411–1423
Core Team R (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Cutter AD (2019) A primer of molecular population genetics. Oxford University Press, Oxford
de Montaigu A, Coupland G (2017) The timing of GIGANTEA expression during day/night cycles varies with the geographical origin of Arabidopsis accessions. Plant Signal Behav 12:e1342026
de Montaigu A et al (2015) Natural diversity in daily rhythms of gene expression contributes to phenotypic variation. Proc Natl Acad Sci U S A 112:905–910
Ding J, Böhlenius H, Rühl MG, Chen P, Sane S, Zambrano JA, Zheng B, Eriksson ME, Nilsson O (2018) GIGANTEA-like genes control seasonal growth cessation in Populus. New Phytol 218:1491–1503
Dormling I (1973) Photoperiodic control of growth and growth cessation in Norway spruce seedlings. IUFRO Division 2, Working Party 2.01.4, Growth Processes, Symposium on Dormancy in Trees. Kornik, Poland, pp 1–16
Doyle JA (2012) Molecular and fossil evidence on the origin of angiosperms. In: Jeanloz R (ed) Annual review of earth and planetary sciences, vol 40. Department of Evolution and Ecology, University of California, Davis, pp 301-326
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Ewing B, Green P (1998) Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier LD, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res 8:175–185
Fischer MC, Foll M, Excoffier L, Heckel G (2011) Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Mol Ecol 20:1450–1462
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Foll M, Fischer MC, Heckel G, Excoffier L (2010) Estimating population structure from AFLP amplification intensity. Mol Ecol 19:4638–4647
Fornara F, de Montaigu A, Coupland G (2010) SnapShot: control of flowering in Arabidopsis. Cell 141:550
González-Martínez SC, Alía R, Gil L (2002) Population genetic structure in a Mediterranean pine (Pinus pinaster Ait.): a comparison of allozyme markers and quantitative traits. Heredity 89:199–206
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Grivet D, Sebastiani F, Alía R, Bataillon T, Torre S, Zabal-Aguirre M, Vendramin GG, González-Martínez SC (2011) Molecular footprints of local adaptation in two Mediterranean conifers. Mol Biol Evol 28:101–116
Grøndahl E, Ehlers BK (2008) Local adaptation to biotic factors: reciprocal transplants of four species associated with aromatic Thymus pulegioides and T. serpyllum. J Ecol 96:981–992
Günther T, Coop G (2013) Robust identification of local adaptation from allele frequencies. Genetics 195:205–220
Gyllenstrand N, Clapham D, Källman T, Lagercrantz U (2007) A Norway spruce FLOWERING LOCUS T homolog is implicated in control of growth rhythm in conifers. Plant Physiol 144:248–257
Hall D, Ma XF, Ingvarsson PK (2011) Adaptive evolution of the Populus tremula photoperiod pathway. Mol Ecol 20:1463–1474
Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F, Bergelson J (2011) Adaptation to climate across the Arabidopsis thaliana genome. Science 334:83–86
Heuertz M, De Paoli E, Källman T et al (2006) Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 174:2095–2105
Holliday JA, Ritland K, Aitken SN (2010) Widespread, ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce (Picea sitchensis). New Phytol 188:501–514
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Jain SK, Bradshaw AD (1966) Evolutionary divergence among adjacent plant populations. I. Evidence and its theoretical analysis. Heredity 21:407–441
Jakobsson A, Dinnetz P (2005) Local adaptation and the effects of isolation and population size—the semelparous perennial Carlina vulgaris as a study case. Evol Ecol 19:449–466
Janes JK et al (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602
Joshi J et al (2001) Local adaptation enhances performance of common plant species. Ecol Lett 4:536–544
Källman T et al (2014) Patterns of nucleotide diversity at photoperiod related genes in Norway spruce [Picea abies (L.) Karst]. PLoS One 9:e95306. https://doi.org/10.1371/journal.pone.0095306
Karlgren A, Gyllenstrand N, Clapham D, Lagercrantz U (2013) FLOWERING LOCUS T/TERMINAL FLOWER1-Like genes affect growth rhythm and bud set in Norway Spruce. Plant Physiol 163:792–803
Keller SR, Levsen N, Olson MS, Tiffin P (2012) Local adaptation in the flowering-time gene network of Balsam poplar, Populus balsamifera L. Mol Biol Evol 29:3143–3152
Kosintsev PA, Lapteva EG, Trofimova SS, Zanina OG, Tikhonov AN, Van der Plicht J (2012) Environmental reconstruction inferred from the intestinal contents of the Yamal baby mammoth Lyuba (Mammuthus primigenius Blumenbach, 1799). Quat Int 255:231–238
Lascoux M, Glémin S, Savolainen O (2016) Local adaptation in plants. In: eLS. Chichester. John Wiley & Sons, Ltd., pp 1–7
Le Corre V, Kremer A (2003) Genetic variability at neutral markers, quantitative trait loci and trait in a subdivided population under selection. Genetics 164:1205–1219
Leimu R, Fischer M (2008) A meta-analysis of local adaptation in plants. PLoS One 3:e4010
Liu L, Adrian J, Pankin A, Hu J, Dong X, Von Korff M, Turck F (2014) Induced and natural variation of promoter length modulates the photoperiodic response of FLOWERING LOCUS T. Nat Commun 5:4558
Lockwood JD, Aleksic JM, Zou J, Wang J, Liu J, Renner SS (2013) A new phylogeny for the genus Picea from plastid, mitochondrial, and nuclear sequences. Mol Phylogenet Evol 69:717–727
Lotterhos KE, Whitlock MC (2015) The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol Ecol 24:1031–1046
Macel M, Lawson CS, Mortimer SR, Smilauerova M, Bischoff A, Crémieux L, Dolezal J, Edwards AR, Lanta V, Bezemer TM, van der Putten W, Igual JM, Rodriguez-Barrueco C, Müller-Schärer H, Steinger T (2007) Climate vs. soil factors in local adaptation of two common plant species. Ecology 88:424–433
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351:652–654
Milesi P et al (2019) Assessing the potential for assisted gene flow using past introduction of Norway spruce in Southern Sweden: local adaptation and genetic basis of quantitative traits in trees. Evol Appl 12:1946–1959 (in press)
Mishra P, Panigrahi KC (2015) Gigantea—an emerging story. Front Plant Sci 6:1–15
Nei M, Li W-H (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A 76:5269–5273
Nicholson G, Smith AV, Jonsson F, Gustafsson O, Stefansson K, Donnelly P (2002) Assessing population differentiation and isolation from single-nucleotide polymorphism data. J R Statist Soc B 64:695–715
Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A, Vicedomini R, Sahlin K, Sherwood E, Elfstrand M, Gramzow L, Holmberg K, Hällman J, Keech O, Klasson L, Koriabine M, Kucukoglu M, Käller M, Luthman J, Lysholm F, Niittylä T, Olson A, Rilakovic N, Ritland C, Rosselló JA, Sena J, Svensson T, Talavera-López C, Theißen G, Tuominen H, Vanneste K, Wu ZQ, Zhang B, Zerbe P, Arvestad L, Bhalerao R, Bohlmann J, Bousquet J, Garcia Gil R, Hvidsten TR, de Jong P, MacKay J, Morgante M, Ritland K, Sundberg B, Thompson SL, van de Peer Y, Andersson B, Nilsson O, Ingvarsson PK, Lundeberg J, Jansson S (2013) The Norway spruce genome sequence and conifer genome evolution. Nature 497:579–584
Oddou-Muratorio S, Davi H (2014) Simulating local adaptation to climate of forest trees with a physio-demo-genetics model. Evol Appl 7:453–467
Opseth L, Holefors A, Rosnes AKR, Lee Y, Olsen JE (2016) FTL2 expression preceding bud set corresponds with timing of bud set in Norway spruce under different light quality treatments. Environ Exp Bot 121:121–131
Pais AL, Whetten RW, Xiang QJ (2017) Ecological genomics of local adaptation in Cornus florida L. by genotyping by sequencing. Ecol Evol 7:441–465
Pavy N, Namroud MC, Gagnon F, Isabel N, Bousquet J (2012) The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity 108:273–284
Pin PA, Nilsson O (2012) The multifaceted roles of FLOWERING LOCUS T in plant development. Plant Cell Environ 35:1742–1755
Pin PA, Benlloch R, Bonnet D, Wremerth-Weich E, Kraft T, Gielen JJL, Nisson O (2010) An antagonistic pair of FT homologs mediates the control of flowering time in sugar beet. Science 330:1397–1400
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Pickrell JK, Coop G (2010) The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol 20:R208–R215
Pyhäjärvi T, Salmela MJ, Savolainen O (2007) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet Genomes 4:247–254
Rand DM, Kann LM (1996) Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol 13:735–748
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES 4th (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci U S A 98:11479–11484
Savolainen O, Pyhäjärvi T, Knürr T (2007) Gene flow and local adaptation in trees. Annu Rev Ecol Evol Syst 38:595–619
Savolainen O et al (2011) Adaptive potential of northernmost tree populations to climate change, with emphasis on Scots pine (Pinus sylvestris L.). J Hered 102:526–536
Savolainen O, Lascoux M, Merilä J (2013) Ecological genomics of local adaptation. Nat Rev Genet 14:807–820
Schwartz S, Balasubramanian S, Warthmann N, Michael TP, Lempe J, Sureshkumar S, Kobayashi Y, Maloof JN, Borevitz JO, Chory J, Weigel D (2009) Cis-regulatory changes at FLOWERING LOCUS T mediate natural variation in flowering responses of Arabidopsis thaliana. Genetics 183:723–732
Stoletzki N, Eyre-Walker A (2011) Estimation of the neutrality index. Mol Biol Evol 28:63–70
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
The 1001 Consortium (2016) Genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166:481–491
Tsuda Y, Chen J, Stocks M, Källman T, Sønstebø JH, Parducci L, Semerikov V, Sperisen C, Politov D, Ronkainen T, Väliranta M, Vendramin GG, Tollefsrud MM, Lascoux M (2016) The extent and meaning of hybridization and introgression between Siberian spruce (Picea obovata) and Norway spruce (Picea abies): cryptic refugia as stepping stones to the west? Mol Ecol 25:2773–2789
Väliranta M, Kaakinen A, Kuhry P, Kultti S, Salonen JS, Seppä H (2011) Scattered late-glacial and early Holocene tree populations as dispersal nuclei for forest development in north-eastern European Russia. J Biogeogr 38:922–932
Velichko AA, Timireva SN, Kremenetski KV, MacDonald GM, Smith LC (2011) West Siberian Plain as a late glacial desert. Quat Int 237:45–53
Vilhjalmsson BJ, Nordborg M (2013) The nature of confounding in genome-wide association studies. Nat Rev Genet 14:1–2
Wang J et al (2018) A major locus controls local adaptation and adaptive life history variation in a perennial plant. Genome Biol 19:72
Watterson GA (1975) On the number of segregating sites in genetic models without recombination. Theor Popul Biol 7:256–276
Yeaman S et al (2016) Convergent local adaptation to climate in distantly related conifers. Science 353:1431–1433
Zhou Y et al (2018) Comparative transcriptomics provides novel insights into the mechanisms of selenium tolerance in the hyperaccumulator plant Cardamine hupingshanensis. Sci Rep 8:2789
Acknowledgments
Open access funding provided by Uppsala University. We thank Vladimir L. Semerikov for his help during the sampling along the Ob River and Nannan Xu for her help with the growth chamber experiments and the sequencing of the samples. We are grateful for the useful comments from the reviewers and the associate editor.
Data Archiving Statement
Sequences, records of height measurements in the growth chamber experiment and FTL2 gene expression, and the data related to the population structure are deposited in OSF repository under https://osf.io/t745f/?view_only=69f09a3966d0459c8fa1bc52ff55e907.
Funding
This study was supported by the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS). Lili Li was supported by the Chinese Scholarship Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Gugerli
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, L., Chen, J. & Lascoux, M. Clinal variation in growth cessation and FTL2 expression in Siberian spruce. Tree Genetics & Genomes 15, 82 (2019). https://doi.org/10.1007/s11295-019-1389-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-019-1389-7