
Abstract
Vibrio natriegens is a fast-growing, non-pathogenic marine bacterium with promising features for biotechnological applications such as high-level recombinant protein production or fast DNA propagation. A remarkable short generation time (< 10 min), robust proteosynthetic activity and versatile metabolism with abilities to utilise wide range of substrates contribute to its establishment as a future industrial platform for fermentation processes operating with high productivity.
D,D-carboxypeptidases are membrane-associated enzymes involved in peptidoglycan biosynthesis and cell wall formation. This study investigates the impact of overexpressed D,D-carboxypeptidases on membrane integrity and the increased leakage of intracellular proteins into the growth medium in V. natriegens. Our findings confirm that co-expression of these enzymes can enhance membrane permeability, thereby facilitating the transport of target proteins into the extracellular environment, without the need for secretion signals, tags, or additional permeabilization methods. Using only a single step IMAC chromatography, we were able to purify AfKatG, MDBP or Taq polymerase in total yields of 117.9 ± 56.0 mg/L, 36.5 ± 12.9 mg/L and 26.5 ± 6.0 mg/L directly from growth medium, respectively. These results demonstrate the feasibility of our V. natriegens based system as a broadly applicable extracellular tag-less recombinant protein producer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prokaryotic host systems such as Escherichia coli or Bacillus subtilis are widely used for protein overproduction and allow to obtain large quantities of recombinant proteins in a short time. When pursuing industrial production, there is a preference for bacterial hosts that are fast-growing, well-established, and cost-effective. Novel prokaryotic platform based on V. natriegens offers many advantages over widely used host systems for protein production at laboratory and industrial scales. V. natriegens is gram-negative, non-pathogenic bacterium with shorter doubling time than E. coli (< 10 min) (Payne et al. 1961). It has a rapid proteosynthesis and exceptionally high biomass specific consumption rate (qs) under aerobic and anaerobic conditions (Hoffart et al. 2017). Moreover, V. natriegens possesses a robust metabolism, utilise various substrates and grows rapidly on low-cost media (Kormanová et al. 2020). Moreover, V. natriegens offers great metabolic capacity for intracellular and potentially extracellular protein production than E. coli (Schwarz et al. 2022).
Extracellular secretion offers several advantages over intracellular production, including simplifying downstream processes and improving the biological activity, solubility, and stability. Additionally, an extracellular production strategy becomes crucial when dealing with the expression of toxic proteins (Yoon et al. 2010). Similar to E. coli, V. natriegens has limited secretion capacity and typically cannot produce recombinant proteins in sufficient quantities directly into the growth medium (Cornelis 2000; Yoon et al. 2010). One of the extracellular production strategies includes a specific translocation across inner cytoplasmatic membrane using a secretion signal and translocation machinery. Especially TSS1 and TSS2 have been widely used for secretion of recombinant proteins in E. coli (Korotkov et al. 2012). However, one of the main drawbacks are a limited capacity of these machineries, weak secretion signal or size of secreted proteins. When the transport mechanism reaches its capacity, proteins have a tendency to form insoluble aggregates. Moreover, the secretion signal typically exhibits a specific secretion affinity when attached to a different recombinant protein, therefore the extensive optimization process is required. In general, large multidomain intracellular proteins may encounter difficulties in crossing the membrane. Therefore, additional approaches are required to enhance outer membrane permeabilization, such as the co-expression of lysis-promoting proteins, the addition of supplements like glycine (Yang et al. 1998), Triton X-100 (Fu 2010) or using leaky L-form mutant strains (Gumpert et al. 1998; Pang et al. 2022). However, these methods are often not suitable for industrial applications and high-density cell production of recombinant proteins.
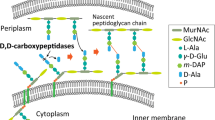
A study by Yang et al. (2019) reported an alternative approach to enhance the permeability of both the inner and outer membrane in E. coli. This approach involves the overexpression of specific membrane-associated D-alanyl-D-alanine-carboxypeptidases (PBPs). Penicillin-binding proteins (PBPs) are proteins associated with the cell membrane that play a crucial role in the biosynthesis of peptidoglycan (PG). Their primary function is to catalyse the final steps in PG biosynthesis. The difference in the number and specificity of PBPs among different bacterial species is the results of specific environment conditions and different PG biosynthesis mechanisms. PBPs could be categorized into two main subclasses: High Molecular Weight (HMW) PBPs and Low Molecular Weight (LMW) PBPs. HMW PBPs consists of multi-domain proteins responsible for PG polymerization and biosynthesis. According to the activity and structure of N-terminal domain, Class A and Class B of HMW PBPs were characterized (Gittins et al. 1994). Class A PBPs are bifunctional proteins which have glycosyltransferase activity responsible for elongation of uncross-linked glycan units and transpeptidase activity of C-terminal domain providing peptide cross-linking between two glycan chains. Class B PBPs plays an important role in the cell morphogenesis and cooperation with other main proteins in the cell cycle. The LMW PBPs, with their D-alanyl-D-alanine carboxypeptidase activity, also play an important role in PG biogenesis and cell wall assembling by maintaining PG crosslinking and stabilization of cell wall structure. However, the LMW PBPs are often not essential for cell viability and growth. Therefore, they were selectively targeted for the generation of deletion mutants, aiming to induce membranes with a certain degree of permeability while minimizing the loss in viability (Denome at al. 1999; Ghosh et al. 2008). The overexpression of certain LMW PBPs could potentially also weaken PG structures of the cell wall and membrane integrity (Yang et al. 2019).
In general, the extracellular secretion approach can offer time and cost-saving benefits. We have designed a simple system for enhancing permeability in V. natriegens to provide alternative approach for extracellular targeting of recombinant proteins without the need for fusion-tags or secretion signals. This system is based solely on the strategy of overexpressing D,D-carboxypeptidases, which promotes the non-selective leakage of proteins into the growth media in V. natriegens.
Materials and methods
Construction of recombinant plasmids
The genes representing D,D-carboxypeptidases PBP4 and PBP5/6 respectively (Table 1) were amplified from V. natriegens ATCC 14,048 genome by PCR with specific primers and inserted into plasmid pRSFDuet (Table 2). Both genes were placed under T5 and T7 promoter resulting in recombinant constructs pRSFDuet-T7-PBP4, pRSFDuet-T7-PBP5/6, pRSFDuet-T5-PBP4 and pRSFDuet-T5-PBP5/6.
Media and culture conditions
LB3 medium (10 g/L peptone, 5 g/L yeast extract, 30 g/L NaCl) was used for cultivation of all V. natriegens strains (V. natriegens Vmax, Synthetic Genomics®, La Jolla, CA and V. natriegens PF, Pfeifer et al. 2019). The cultivations were carried on in the shake flasks at 37 °C on a rotary shaker (200 rpm). The expression of recombinant protein was induced by the addition of 1 mM IPTG. At the point of induction, bacterial cultures (AfKatG and MDBP expression) were also supplemented with hemin to a final concentration of 0.005%. BHI medium (37 g/L) (Sigma) supplemented with v2 salts (204 mM NaCl, 4.2 mM KCl, and 23.14 mM MgCl2) was used for preparation of electrocompetent cells.
DCW determination
For the determination of dry cell weight (DCW), 1 ml sample of bacterial culture was harvested by centrifuging at 10,000 x g for 10 min at 4 °C. The resulting pellet was then dried at 95 °C until a constant weight was achieved.
Bacterial transformation
V. natriegens strains were transformed using a modified electroporation protocol (Weinstock et al. 2016). V. natriegens cells were grown in BHIv2 medium to OD600 = 0.5. Bacterial culture was incubated on ice for 15 min. Cells were harvested at 4305 x g at 4 °C. Bacterial pellet was washed with electroporation buffer (680 mM sucrose, 7 mM K2HPO4, pH 7.2) and centrifuged at 4305 x g at 4 °C for total of three times. The pellet was resuspended in residual electroporation buffer and diluted with additional buffer to reach optical density value at 600 nm within 18–20. Plasmid DNA (50–200 ng) was added to cells, gently mixed, and transferred to a chilled electroporation cuvette (0.1 cm gap, Bio-Rad). Transformation was carried on with following parameters: 800 V, 25 25 μF, 200 Ω using Gene Pulser™ (Bio-Rad). Cells were immediately incubated in pre-heated recovery LB3 medium supplemented with 680 mM sucrose and shaken at 28 °C for at least 2 h.
GFP assay
V. natriegens strains, containing prepared recombinant plasmids, were cultivated in LB3 medium at 37 °C overnight. Shake flasks (50 ml) with fresh LB3 medium were inoculated to final concentration of 1.0% (v/v) for expression of recombinant GFP. After reaching OD600 0.5, GFP expression was induced by adding ITPG to a final concentration of 1 mM. Fluorescence intensity was measured in black 96-well plate using Varioskan Flash (Thermo Scientific). Excitation and emission wavelengths were 488 and 533 nm, respectively.
Enzyme activity assay
Specific catalase activity was determined as decrease of an absorbance at 240 nm in phosphate buffer pH 6.0 with 10 mM H2O2 as substrate. The kinetics was measured by UV-Vis Spectrophotometer (UV-1800, SHIMADZU) for 2 min with 1 s interval for absorbance measurement. Results were recorded using UV Probe Ver. 2.42 software.
Specific peroxidase activity was determined as increase of absorbance at 405 nm in phosphate buffer pH 4.5 with 2 mM H2O2 and 0.9 mM ATBS as a substrate. The kinetics were measured in the same manner as in the case of catalase activity.
Outer membrane assay
N-Phenyl-α-naphthylamine (NPN) was used for determination of outer membrane permeability (Loh et al. 1984). Cells were harvested by centrifugation at room temperature at 1000 x g for 10 min. Pellet was washed in 5 mM HEPES pH 7.2 and cells were diluted at final OD600 = 0.5. NPN solution was added to the cell suspension to the final concentration of 40 μM. Samples were immediately measured in a black 96-well plate using Varioskan Flash (Thermo Scientific). Measurements were performed before induction (0 min) and after induction (360 min). The fluorescence intensity was measured at excitation and emission wavelengths of 350 and 420 nm, respectively.
Inner membrane assay
Propidium iodide (PI) was used for determination of inner membrane permeability (Ma et al. 2020). Cells were harvested by centrifugation at 8935 x g for 10 min after induction of D,D-carboxypeptidase overexpression and the supernatant was carefully removed. Cells were washed in the wash buffer (5 mM HEPES, 5 mM glucose pH 7.2). After the second round of centrifugation at 8935 x g cells were resuspended in the wash buffer. PI was added to the final concentration of 5 μM and incubated in the dark, at room temperature for 30 min. Measurements were performed before induction (0 min) and after induction (240 min). The fluorescence intensity was measured using Varioskan Flash (Thermo Scientific) at excitation wavelength 535 nm and emission wavelength 617 nm in the black 96-well plate.
SDS-PAGE analysis
Samples for SDS-PAGE analysis of cells containing targeted proteins were harvested by centrifugation, resuspended in TE buffer, loading buffer, and incubated at 95 °C for 10 min. Prepared samples were analysed in 12% SDS-polyacrylamide gel (Laemmli, 1974).
An internal control sample for Western blot analysis was prepared using the same procedure as the other samples. The internal control consisted of a sample obtained from a 24-hour intracellular expression of Taq polymerase, following the exact conditions applied to the tested samples.
For protein sample preparation from growth medium, 1 ml of medium was deprived of bacterial cells by previous centrifugation and precipitated using trichloroacetic acid (TCA)/acetone. A volume of 250 μl TCA was added and incubated at 4 °C for 10 min. The sample was centrifuged at 21,952 x g for 5 min, pellet was washed with 200 μl acetone twice and dried at 95 °C for 5–10 min. Samples for SDS-PAGE were prepared as described for cell protein samples.
Sample preparation for affinity chromatography
Bacterial cells were harvested at 7650 x g at 4 °C for 20 min and resuspended in equilibration buffer (TrisHCl pH 8.0, 0.5 M NaCl). Cells were disrupted by ultrasound using homogenizer Sonopuls HD3200 with KE 76 probe (Bandelin). The cell debris was removed by centrifugation at 7650 x g at 4 °C. Soluble fraction was stored on ice.
For protein purification from the growth media, bacterial cells were also harvested at 7650 x g at 4 °C for 20 min. Medium was carefully separated from cell pellet and diluted with equilibration buffer in ratio 1:1. Prepared sample was stored on ice.
Affinity chromatography
Recombinant proteins (AfKatG, MDBP, Taq, MutTaq) were expressed in fusion with His-tag sequence and afterwards purified using affinity chromatography on ÄKTA Avant 25 FPLC system (GE Healthcare) with 1 ml HisTrap HP (GE Healthcare). At the beginning of the purification run, the column was washed with 5 column volumes (5 CV) of equilibration buffer (50 mM TrisHCl, pH 8.0, 0.5 M NaCl). Sample was loaded onto column using the pump at speed of 0.1-1 CV/min. Afterwards, the column was washed with equilibration buffer (10 CV) with 5% addition of elution buffer (50 mM Tris-HCl pH 8.0, 0.5 M NaCl, 0.5 M imidazole) to remove non-bound and non-specifically bound proteins. Elution buffer with 0.5 M imidazole was used to release bound His-tagged proteins. Samples were taken automatically by the fraction collector based on predefined parameters.
Western blot analyses
Protein samples separated on SDS-PAGE gels were transferred to the polyvinylidene fluoride (PVDF) membrane. The semi-dry transfer was performed in Tris-glycine buffer with SDS and isopropanol using Biometra Flashblot (Analytik Jena GmbH). Parameters of the analysis were set to 15 V, 250 mA for 45 min. Subsequently, the PVDF membrane was incubated in specific antibody solution (mouse anti-His) (Invitrogen) with dilution of 1:1000 in a 1% skim milk solution at 4 °C overnight. Further, the membrane was washed in goat anti-mouse secondary antibody (1:10 000) in 1% skim milk solution (Goat Anti-Mouse IgG, Sigma Aldrich) for 1 h at room temperature. For signal visualisation, the membrane was washed in chemiluminescence solution using Clarity Max Western ECL Substrate kit (Bio-Rad) according to the protocol and signal was detected using ImageQuant LAS 500 chemiluminescence CCD camera (GE Healthcare).
Statistical analyses
All experiments were prepared in triplicates. Data are displayed as a mean with standard deviations (SD). For statistical analysis the student t test was used, and a P value of < 0.05 was considered as statistically significant.
Results
Effect of D,D-carboxypeptidases PBP4 or PBP5/6 overexpression on V. natriegens biomass formation
For purpose of this work, we have applied two V. natriegens strains for the determination of D,D-carboxypeptidase overexpression impact on culture growth and biomass formation. V. natriegens Vmax host strain (Synthetic Genomics®; La Jolla, California) possesses an inserted T7 polymerase cassette and is compatible with the T7-based series of expression vectors. V. natriegens PF (Prophage-free), prepared in the work Pfeifer et al. (2019), contains deletions of predicted prophage loci. These deletions have a significant effect on robustness and production stability of the strain (Pfeifer et al. 2019). However, V. natriegens PF is lacking T7-polymerase cassette, therefore we applied vectors with a hybrid T5-lac promoter for overexpression of PBPs and other target proteins. In case of V. natriegens Vmax, we have applied T7 promoter and T7 expression system as a useful alternative for production of recombinant proteins.
First, we investigated the influence of D,D-carboxypeptidases PBP4 or PBP5/6 overexpression on V. natriegens growth. The decrease in a biomass formation was not statistically significant in all tested cultivations. However, the overexpression of PBP5/6 caused a stronger inhibition of biomass formation when overexpressed in both strains (Fig. 1). Moreover, we observed no significant impact on specific growth rate in V. natriegens Vmax or PF cultivations when PBP4 or PBP5/6 were overexpressed (See supplementary material). We also observed the increase of protein abundance in growth medium when PBP4 and PBP5/6 overexpressed in both V. natriegens strains. The strongest leak of intracellular protein was detected after 24 h of cultivation when compared to a control (See supplementary material).

Determination of PBP4 or PBP5/6 D,D-carboxypeptidase overexpression on cell growth and biomass formation (dashed lines). Growth curves for V. natriegens Vmax and PF strains were determined according to DCW increase. D,D-carboxypeptidase overexpression was not present in control cultivations (Vmax and PF – solid lines). PF – V. natriegens Prophage-free; Vmax – V. natriegens Vmax; DCW - dry cell weight
Determination of outer and inner membrane permeability in V. natriegens cells after D,D-carboxypeptidase PBP4 or PBP5/6 overexpression
NPN was used to detect outer membrane permeability. NPN fluorescence is in an aqueous environment weak, however it is highly increased in a nonpolar or hydrophobic environment. Overexpression of PBP5/6 caused an increase in fluorescent intensity in V. natriegens Vmax and V. natriegens PF in comparison with control (Fig. 2A). After PBP5/6 overexpression, we detected 2.8-fold increase of the fluorescence intensity in V. natriegens Vmax compared to control. Overexpression of PBP4 has no significant effect on outer cell membrane permeability in both V. natriegens strains (Fig. 2A).

A - Outer membrane permeability of V. natriegens strains was determined by a fluorescence increase resulting from NPN uptake into the hydrophobic area of outer membrane. B - Inner membrane permeability of V. natriegens strains was determined by a fluorescence increase resulting from PI uptake into intracellular area of the cell. The cultivation without overexpression of PBP4 or PBP5/6 D,D-carboxypeptidases was used as a control. A p-value of less than 0.05 was considered statistically significant. PF – V. natriegens Prophage-free, Vmax – V. natriegens Vmax
PI is a red fluorescent nucleic acid dye and was used to detect inner membrane permeability. Overexpression of D,D-carboxypeptidase PBP5/6 caused the significant increase in PI fluorescent intensity in V. natriegens Vmax (by 3.2-fold) and in V. natriegens PF (by 2.5-fold) (Fig. 2B). Therefore, we assume that the overexpression of D,D-carboxypeptidase PBP5/6 might improve inner membrane permeability of V. natriegens strains.
Overexpression of D,D-carboxypeptidase and its effect on extracellular GFP transport
We also investigated an effect of co-expression of D,D-carboxypeptidases PBP4 or PBP5/6 with GFP and its influence on GFP leak into a growth media. We have not observed any significant differences between intracellular fluorescence intensity in V. natriegens Vmax (Fig. 3A) or V. natriegens PF (Fig. 3B). However, the fluorescence intensity in growth medium was increased in case of the overexpression both carboxypeptidases PBP4 and PBP5/6 in V. natriegens Vmax and V. natriegens PF. After PBP4 overexpression the fluorescence intensity increased by fold of 0.9 in V. natriegens Vmax and 4.5-fold in V. natriegens PF compared to control. In case of PBP5/6 overexpression the fluorescence intensity increased by fold of 6.4 in V. natriegens Vmax and 2.1-fold in V. natriegens PF compared to control.

Effect of the D,D-carboxypeptidase PBP4 or PBP5/6 overexpression under T7-promoter on intracellular production of GFP as well as its extracellular transport into growth media in V. natriegens Vmax and PF A – Fluorescence intensity of intracellular and extracellular GFP with PBP4 or PBP5/6 co-expression after induction with 1 mM IPTG in V. natriegens Vmax; B – Fluorescence intensity of intracellular and extracellular GFP in medium after PBP4 or PBP5/6 co-expression after induction with 1 mM IPTG in V. natriegens PF; Control cultivations were lacking permeabilization plasmids for PBP4 and PBP5/6 overexpression. IP – Intracellular protein; EP – Extracellular protein; GFP - Green fluorescent protein; A p-value less than 0.05 was considered statistically significant. PF – V. natriegens Prophage-free; Vmax – V. natriegens Vmax
Overexpression of PBP5/6 enzyme and its effect on AfKatG extracellular transport
Thermostable catalase-peroxidase (AfKatG) is an enzyme originally isolated from hyperthermofillic archaeon Archeoglobus fulgidus (Beeder et al. 1994). It can be found in hydrothermal environment such as hot springs with the approximate temperature of 70–80 °C. Its catalase and peroxidase activity towards wide spectrum of substrates was in detail described. AfKatG is an interesting enzyme for a variety of industrial applications. From the structural point of view, it is a homodimer with subunit molecular mass of 85 kDa and contains binding site for a prosthetic group (ferric heme) in the stoichiometric ratio of 0.25 heme per subunit, crucial for its enzyme activities (Kengen et al. 2001).
AfKatG was expressed under T5-lac promoter in V. natriegens PF to confirm its intracellular production and activity (Fig. 4A). Afterwards, AfKatG was used as a model protein for demonstration of intracellular protein leak into the growth medium using PBP5/6 D,D-carboxypeptidase co-expression in V. natriegens PF. When PBP5/6 co-expression was present, no significant decrease occurred in the intracellular production of AfKatG. However, a significant increase in extracellular AfKatG abundance occurred when the D,D-carboxypeptidase PBP5/6 was co-expressed (Fig. 4A). After purification from constant volume of growth medium using IMAC (at the same conditions) we were able to reach yields of 117.9 ± 56.0 mg/L when PBP5/6 co-expressed (Table 3). In control cultivation, the total gained yield of AfKatG purified from growth medium was 3.2 ± 1.3.

A - SDS-PAGE analysis of AfKatG production and its transport into growth medium in V. natriegens PF with co-expression of PBP5/6 under T5-lac promoter; B – SDS-PAGE analysis of AfKatG after IMAC purification from growth media. MS – protein molecular weight standard; IP – intracellular protein; EP – extracellular protein; FT – flow through; W- wash fraction; E – elution fraction; PF – V. natriegens Prophage-free; the numbering of lanes on SDS-PAGE or Western blot corresponds to the time in hours elapsed after induction
Catalase and peroxidase activity was measured to confirm catalytic activity of AfKatG. No significant difference in catalase or peroxidase activity was detected when isolated from medium, cells or permeabilized cells (Table 3).
Overexpression of PBP5/6 enzyme and its effect on MDBP peroxidase extracellular transport
MDPB (manganese-dependent bacterial peroxidase) enzyme belongs to the peroxidase family which was previously identified through bioinformatics analysis. The primary structure of the protein involves GXXDG motive which is specific for peroxidases and is also binding site for a prosthetic group (ferric heme). This enzyme is characterized by its ability to degrade lignin and cellulose, making it highly promising for various biotechnological applications.
MDBP was produced under T5-lac promoter in V. natriegens PF to investigate a co-expression effect of PBP5/6 and its transfer into growth medium (Fig. 5A). The level of intracellular production based on densitometric analysis was after 24 h ± 21% of TCP in control and ± 19% of TCP when PBP5/6 co-expressed. The soluble fraction of MDBP was calculated at the level of ± 31% and ± 33%, with and without co-expression of PBP5/6, respectively. Further, we also purified MDBP from cell fraction and medium using IMAC chromatography (Fig. 5B). We detected a minimal effect on MDBP intracellular production when PBP5/6 co-expressed. However, after purification of MDBP directly from growth medium, we detected a significant increase of MDBP in elution sample compared to control cultivations (Table 4). Total purified yield of MDBP from medium using one-step IMAC was 36.5 ± 12.9 mg/L resulting in increase compared to the control.

A - SDS-PAGE analysis of MDBP production and its extracellular transport into growth medium in V. natriegens PF with co-expression of PBP5/6 under T5-lac promoter; B – SDS-PAGE analysis of MDBP production after IMAC purification from growth medium. Control cultivation was lacking plasmid for D,D-carboxypeptidase overexpression. MS – protein molecular weight standard; IP – intracellular protein; EP – extracellular protein; the numbering of lanes on SDS-PAGE or Western blot corresponds to the time in hours elapsed after induction
Peroxidase activity was measured to confirm its catalytic activity (Table 4). No significant difference in peroxidase activity of MDBP isolated from medium, cells or permeabilized cells was observed.
Overexpression of PBP5/6 enzyme and its effect on Taq and MutTaq polymerase extracellular transport
DNA polymerases are often used for various genetic engineering techniques, including DNA cloning, mutagenesis, sequencing, and DNA labelling. One of the most widely used polymerases is a thermostable Taq polymerase from Thermus aquaticus. Protein engineering techniques were used to generate mutant enzymes, such as a cold-sensitive mutant of Taq polymerase or mutant Taq polymerase with a resistance to various PCR inhibitors including plasma, blood, hemoglobin or lactoferrin (Kermekchiev et al. 2003, 2009). The design of novel mutant enzymes could provide a promising alternative to the currently available commercial polymerases.
In this study, we expressed the native Taq polymerase (Fig. 6A) and a mutant Taq (MutTaq) polymerase (Fig. 7A) with size of 94 kDa. The expression was carried out under the T7 promoter in V. natriegens Vmax. We compared the extracellular production of these enzymes using PBP5/6 co-expression approach to a control intracellular production (without PBP5/6 overexpression).

A - SDS-PAGE analysis of Taq polymerase production and its extracellular transport into growth medium in V. natriegens Vmax with co-expression of PBP5/6 under T7 promoter; B – Western blot analysis of Taq polymerase production after IMAC purification from growth medium. Control cultivations were lacking plasmid for PBP5/6 overexpression. MS – protein molecular weight standard; FT – flow through; W- wash fraction; E – elution fraction, IP – intracellular protein; EP – extracellular protein; Vmax – V. natriegens Vmax; the numbering of lanes on SDS-PAGE or Western blot corresponds to the time in hours elapsed after induction

A - SDS-PAGE analysis of MutTaq polymerase production and its extracellular transport into growth media in V. natriegens Vmax with co-expression of PBP5/6 under T7 promoter; B – Western blot analysis of MutTaq polymerase production after IMAC purification from growth medium. Control cultivations were lacking plasmid for D,D-carboxypeptidase PBP5/6 overexpression. MS – protein molecular weight standard; P – insoluble fraction; S – soluble fraction of MutTaq polymerase after 24 h intracellular production; FT – flow through; W- wash fraction; E – elution fraction, IP – intracellular protein; EP – extracellular protein; Vmax – V. natriegens Vmax; the numbering of lanes on SDS-PAGE or Western blot corresponds to the time in hours elapsed after induction
The co-expression of PBP5/6 enhanced the leak of intracellular proteins including already overexpressed wild type Taq polymerase into growth medium. Following affinity chromatography, the total yield of purified Taq polymerase directly obtained from the growth medium was approximately 26.5 ± 6.0 mg/L (Fig. 6B).
To enhance the beneficial properties of Taq polymerase for future PCR applications (ongoing research), specific mutations were introduced into its DNA sequence. However, these introduced mutations had a negative effect on its solubility (Fig. 7A). Mutated Taq polymerase (MutTaq) was used as a model protein with a low solubility for demonstration of PBP5/6 co-expression strategy (Fig. 7A and B). The intracellular production of MutTaq polymerase after 24 h was at level of 11% TCP and 8.7% TCP when PBP5/6 co-expressed. The soluble intracellular fraction was not observed after Western blot analysis (Fig. 7B). The significant higher amount of MutTaq polymerase was observed only when purified from growth medium with co-expression of PBP5/6. We verified this also by Western blot analysis (Fig. 7B).
Both purified Taq and MutTaq polymerases were lacking their enzymatic activity (See supplementary material). We incorporated an ammonium sulfate precipitation step in the downstream processing of the polymerases to eliminate any residual nucleic acids that might potentially affect the activity of these enzymes (Pluthero 1993). However, our attempts to obtain an active sample of Taq or MutTaq polymerase were unsuccessful, indicating the need for further optimization (Table 5).
Discussion
We investigated the impact of overexpressing D,D-carboxypeptidases PBP4 and PBP5/6 on the transport of model proteins into the growth media, as well as their influence on both outer and inner membrane permeability, biomass formation, and the potential for recombinant protein production. As previously mentioned, PBPs play an important role in PG biosynthesis, maturation, and maintaining the robustness of surface structures of the bacterial cell. We hypothesized that the homologues of D,D-carboxypeptidases within the LMW PBPs group, specific for V. natriegens strains, could affect the robustness and stability of surface structures without causing significant negative effects on viability or overall recombinant protein production. We focused especially on LMW PBPs because HMW PBPs are often crucial for cell viability and their deletion could result in the range of negative effects. For instance, the deficiency of either PBP1 or PBP2 of HMW PBPs in E. coli slows the growth, disrupts surface structures, changes the morphology of bacterial cell, and eventually results in complete cell lysis (Yousif et al. 1985; Suzuki 1978). However, the LMW PBP are often not essential for cell viability and survival. Extensive morphological aberrations and crucial dysfunctions were observed just in E. coli mutants lacking multiple LMW PBPs (Nelson et al. 2002). The deletion of the LMW PBP5 homologue, possessing D,D-carboxypeptidase activity, primarily affects cell shape and membrane stability in E. coli, as well as in other bacterial species such as Bacillus subtilis, Listeria monocytogenes or Vibrio parahaemolyticus (Korsak et al. 2005; Atrih et al. 1999; Hung et al. 2013). LMW PBPs with D,D-carboxypeptidase activity are crucial for removing terminal D-alanine from the pentapeptide chain, which facilitates and enhances PG crosslinking. The deletion or overexpression of these enzymes has a potential to cause an imbalance in PG biosynthesis, crosslinking, or bacterial cell stability. This can lead to greater leakage of intracellular proteins into the growth medium in E. coli (Hu et al. 2019; Yang et al. 2019).
We used the V. natriegens PF strain as a host for the overexpression of D,D-carboxypeptidases PBP4 and PBP5/6 and compared its growth pattern with the commercial host strain V. natriegens Vmax. The overexpression of D,D-carboxypeptidases PBP4 and PBP5/6 in both V. natriegens strains had no significant impact on biomass formation during cultivation, as we assumed based on previous research (Korsak et al. 2005; Atrih et al. 1999; Hung et al. 2013).
V. natriegens PF strain lacks two prophage loci in its genome and has potential advantages in terms of robustness against DNA damage and hypo-osmotic stress. Furthermore, this strain has demonstrated the capability to outcompete wild-type strains in growth and biomass formation (Pfeifer, 2019). These characteristics could be particularly useful in the context of D,D-carboxypeptidase overexpression, as they help minimize any negative effects on biomass production, culture viability, or growth inhibition. After 24 h of cultivation, we observed only a slight decrease in biomass when PBP5/6 was overexpressed in both V. natriegens strains. The growth curves were similar when D,D-carboxypeptidases were co-expressed, either in V. natriegens Vmax under the T7 promoter or in V. natriegens PF under the T5-lac promoter. No significant effect on biomass formation was observed when using V. natriegens PF strain. Therefore, we decided to use both strains equally as alternative host platforms (Fig. 1).
Next, we have analysed the outer and inner membrane permeability in both V. natriegens strains. After overexpression of LMW PBP5/6, a noticeable increase in fluorescence intensity of NPN was observed after 360 min of incubation, both in V. natriegens PF and V. natriegens Vmax (Fig. 2). This suggests the potential disruption of the outer membrane, facilitating the entry of NPN molecules into the hydrophobic periplasm. We also investigated the complexity of the inner membrane using PI dye, since it can only label bacteria with highly disrupted inner membrane. A significant PI fluorescence was observed after PBP5/6 overexpression in both host strains after 280 min of cultivation, and after 360 min of cultivation only in V. natriegens PF (Fig. 2). The observed increase in inner and outer membrane permeability might be the result of PG formation issues or disruption of the cell membrane.
The analysis of SDS-PAGE revealed the increase in the overall abundance of total proteins in the growth medium after induction of D,D-carboxypeptidase overexpression. The overall abundance of proteins in the medium was highest after 24 h of cultivation in both V. natriegens Vmax and V. natriegens PF strains (See supplementary material). To validate this observation, we performed co-expression experiments involving a model GFP protein with each D,D-carboxypeptidase in V. natriegens PF and V. natriegens Vmax. No significant change in fluorescence intensity was observed for intracellular production (Fig. 3). However, a significant increase in fluorescence intensity was observed in the growth medium compared to the control without co-expression of D,D-carboxypeptidases, particularly after 24 h of cultivation (Fig. 3). Therefore, we performed all subsequent cultivations within a 24-hour timeframe to achieve the highest possible abundance of intracellular model proteins in growth medium.
Subsequently, we performed a co-expression of PBP5/6 D,D-carboxypeptidase in V. natriegens PF to produce the heme protein AfKatG directly into the growth medium. We were able to achieve significantly higher yields of AfKatG in medium compared to the extracellular control of AfKatG without PBP5/6 co-expression. The overall production of AfKatG from medium reached a level of 117.9 ± 56.0 mg/L, which was significantly higher than the control without co-expression of PBP5/6. This alternative approach offers additional advantages by simplifying downstream processes and eliminating the need for homogenization steps. We also observed a small amount of AfKatG in the extracellular fraction of control culture after 24 h. In studies involving Gram-negative bacteria, centrifugation forces exceeding 5000 x g can result in cell disruption or the presence of the protein in medium can be simply the result of natural cell death and disruption during the cultivation cycle. For all downstream processes, we used a protocol using a higher centrifugation force (21,952 x g), which could potentially cause an additional spontaneous lysis and release of AfKatG from cells. However, the same protocol was consistently applied for preparation of all samples including the control (Pembrey et al. 1999).
After 24 h of cultivation, the intracellular abundance of AfKatG decreased by 16% when D,D-carboxypeptidase PBP5/6 was co-expressed, potentially due to the transport of AfKatG into the growth medium. However, no significant change in overall AfKatG concentration from soluble intracellular fraction was detected. This could be due to the shift of the solubility ratio because of lower protein concentration in the cell (due to intracellular protein leakage). Thus, the originally insoluble fraction could in this case replace the AfKatG already transported into the growth medium. The reduction of cellular protein concentration could enhance folding and solubility of protein. Previous studies have reported methods for reducing cellular protein abundance such as using low induction levels of inductor or weak promoter for solubility increase (Sørensen et al. 2005). This explanation was also further supported by densitometric analysis of the soluble fraction, indicating a change in the solubility ratio when comparing AfKatG production with and without D,D-carboxypeptidase PBP5/6 co-expression.
We have further evaluated the system by expressing MDBP heme peroxidase. We observed an increase in overall MDBP extracellular yields by fold of 6.8 when PBP5/6 co-expressed in V. natriegens PF compared to the control (Fig. 5). No significant changes in intracellular production were detected due to solubility shift, similar to AfKatG production.
At the time of induction, all AfKatG and MDBP cultures were supplemented with hemin as the source of prosthetic heme group, which is crucial for maintaining its enzymatic activity. In general, standard laboratory strains have limited capacity to uptake heme from the extracellular environment. Different approaches have been designed to overcome this issue and increase heme content in cells (Varnado et al., 2004). We assumed that permeabilization of bacterial membranes or extracellular production could have a positive impact on homogeneous incorporation of heme into the active site of protein and therefore ensure high activity. For instance, E. coli strain RP523 with introduced uncharacterized permeability mutations was heme-permeable when grown on heme supplemented medium (Li et al. 1988). However, no significant differences were observed in the catalase or peroxidase activity of AfKatG and the peroxidase activity of MDBP in permeabilized cells or samples isolated directly from growth medium (Tables 3 and 4). Thus, this approach may provide an alternative for production of other heme proteins with homogeneous heme content and activity.
We also evaluated the capability of our system by expressing other model proteins such as DNA polymerases. Native thermostable DNA polymerases are usually expressed at low levels by thermophilic bacteria. E. coli or baculovirus production systems were used as hosts for production of active forms of several DNA polymerases. Pfu, Pwo and Taq were produced in E. coli at expression levels of approximately 24 mg/L, 26.6 mg/L and 95 mg/L, respectively (Dabrowski et al., 1998; Engelke et al. 1990). Pfu polymerase was also produced into growth medium using Spodoptera frugiperda cells (sf-9) and Trichoplusia ni cells (hi-5) with final yields of 100 mg/L and 134 mg/L, respectively (Mroczkowski et al. 1994). However, despite their ability to achieve higher production quantities, baculovirus systems are more expensive and more challenging to manipulate compared to bacterial systems. We have used V. natriegens Vmax for the production of wild type Taq polymerase into the growth medium, achieving final yields of 26.5 ± 6.0 mg/L (Table 5). The abundance of the soluble MutTaq polymerase in intracellular fraction was significantly lower compared to the wild-type enzyme when expressed in both V. natriegens strains. However, MutTaq polymerase was an interesting model protein for the application of the PBP5/6 co-expression system due to its high insolubility when produced intracellularly in V. natriegens Vmax (Obr. 7) or in E. coli (data not shown). The intracellular fraction of MutTaq polymerase without D,D-carboxypeptidase PBP5/6 remained insoluble, probably due to high protein concentration. However, we have purified approximately 10.4 ± 5.6 mg/L of MutTaq polymerase directly from medium using the PBP5/6 co-expression strategy. However, both Taq and MutTaq polymerase lacked enzyme activity when purified from the intracellular fraction or directly from the growth medium (See supplementary material). The loss of the polymerisation activity could be the result of chosen method for protein precipitation, protein degradation, or longer timeframe of cultivation (24 h), which is however necessary to reach sufficient yield of model proteins in the growth medium. This limitation could indicate a drawback of our approach.
Targeting recombinant proteins into the growth medium offers several important advantages over intracellular strategies. When a protein is transported directly into growth medium, it remains in its soluble form and reduces negative effects on the bacterial culture and overall production. This approach simplifies the purification process since no cell disruption is required and overall number of purification steps can be reduced while maintaining the same level of product purity (Quax 1997). The downstream process often includes many purification steps to reach the highest purity, resulting in significant loss of valuable material. Production of recombinant proteins directly into growth medium is one of the strategies to reduce the number of required purification steps and overall process costs (Yoon et al. 2010).
In this study, we demonstrated the effect of D,D-carboxypeptidase PBP5/6 overexpression on extracellular leak of model proteins, such as GFP, AfKatG, MDBP and Taq/MutTaq polymerase into the growth medium. As we assumed, co-expression of D,D-carboxypeptidase had a positive impact not just on the abundance of model protein in growth medium but also on purity of these model proteins after chromatography when purified directly from medium. This procedure can be highly beneficial for downstream process optimization, both at laboratory scale and in high-level protein production.
Conclusions
The approach of D,D-carboxypeptidase overexpression could provide the access for intracellular proteins into growth medium without the use of specific secretion signals, fusion tags, additional supplements, or overexpression of secretory machinery and without extensive loss of production capacity of the cell. Nonspecific leakage of intracellular proteins enhanced by overexpression of D,D-carboxypeptidases PBP4 and especially PBP5/6, without significant culture deprivation, could provide a useful way to transport recombinant proteins into media in V. natriegens. Overexpression of D,D-carboxypeptidase PBP5/6 had a positive impact on the transport of intracellular proteins such as GFP, AfKatG, MDBP or Taq polymerase (MutTaq) into growth medium. This simple production strategy could also provide acceleration of downstream processes, reduction of overall production costs, and a promising alternative for production of recombinant proteins with high production rate as well as low solubility in V. natriegens strains.
Data Availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).
References
Atrih A, Bacher G, Allmaier G, Williamson MP, Foster SJ (1999) Analysis of peptidoglycan structure from vegetative cells of Bacillus subtilis 168 and role of PBP 5 in peptidoglycan maturation. J Bacteriol 181:3956–3966
Beeder J, Nilsen RK, Rosnes JT, Torsvik T, Lien T (1994) Archaeoglobus fulgidus isolated from Hot North Sea Oil Field Waters. Appl Environ Microbiol 60:1227–1231
Cornelis P (2000) Expressing genes in different Escherichia coli compartments. Curr Opin Biotechnol 11:450–454
Dabrowski S, Kur J (1998) Cloning and expression in Escherichia coli of the recombinant his-tagged DNA polymerases from Pyrococcus furiosus and Pyrococcus woesei. Protein Exp Purif 14(1):131
Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD (1999) Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol 181(13):3981–3993
Engelke DR, Krikos A, Bruck ME, Ginsburg D (1990) Purification of Thermus aquaticus DNA polymerase expressed in Escherichia coli. Anal Biochem 191(2):396
Fu X-Y (2010) Extracellular accumulation of recombinant protein by Escherichia coli in a defined medium. Appl Microbiol Biotechnol 88:75–86
Ghosh AS, Chowdhury C, Nelson DE (2008) Physiological functions of d-alanine carboxypeptidases in Escherichia coli. Trends Microbiol 16:309–317
Gittins JR, Phoenix DA, Pratt JM (1994) Multiple mechanisms of membrane anchoring of 13 Escherichia coli penicillin-binding proteins. FEMS Microbiol Rev 13:1–12
Gumpert J, Hoischen C (1998) Use of cell wall-less bacteria (L-forms) for efficient expression and secretion of heterologous gene products. Curr Opin Biotechnol 9:506–509
Hoffart E, Grenz S, Lange J, Nitschel R, Müller F, Schwentner A, Feith A, Lenfers-Lücker M, Takors R, Blombach B (2017) High substrate uptake Rates Empower Vibrio natriegens as production host for Industrial Biotechnology. Appl Environ Microbiol 83:01614–01617
Hu J, Lu X, Wang H, Wang F, Zhao Y, Shen W, Yang H, Chen X (2019) Enhancing extracellular protein production in Escherichia coli by deleting the d-alanyl-d-alanine carboxypeptidase gene dacC. Eng Life Sci 19(4):270–278
Hung WC, Jane WN, Wong HC (2013) Association of a D-alanyl-D-alanine carboxypeptidase gene with the formation of aberrantly shaped cells during the induction of viable but nonculturable Vibrio parahaemolyticus. Appl Environ Microbiol 79:7305–7312
Kengen SW, Bikker FJ, Hagen WR, de Vos WM, Van der Oost J (2001) Characterization of a catalase-peroxidase from the hyperthermophilic archaeon Archaeoglobus fulgidus. Extremophiles 5:323–332
Kermekchiev MB, Tzekov A, Barnes WM (2003) Cold-sensitive mutants of taq DNA polymerase provide a hot start for PCR. Nucleic Acids Res 31:6139–6147
Kermekchiev MB, Kirilova LI, Vail EE, Barnes WM (2009) Mutants of taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res 37:40
Kormanová Ľ, Rybecká S, Levarski Z, Struhárňanská E, Levarská L, Blaško J, Turňa J, Stuchlík S (2020) Comparison of simple expression procedures in novel expression host Vibrio natriegens and established Escherichia coli system. J Biotechnol 321:57–67
Korotkov KV, Sandkvist M, Hol WGJ (2012) The type II secretion system: biogenesis, molecular architecture and mechanism. Nat Rev Microbiol 10:336–351
Korsak D, Vollmer W, Markiewicz Z (2005) Listeria monocytogenes lacking penicillin-binding protein 5 (PBP5) produces a thicker cell wall. FEMS Microbiol Lett 251:281–288
Laemmli UK, Paulson JR, Hitchins V (1974) Maturation of the head of bacteriophage T4. A possible DNA packaging mechanism: in vitro cleavage of the head proteins and the structure of the core of the polyhead. J Supramol Struct 2:276–301
Li JM, Umanoff H, Proenca R, Russell CS, Cosloy SD (1988) J Bacteriol 170:1021–1025
Loh B, Grant C, Hancock R (1984) Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob Agents Chemother 26:546–551
Ma B, Fang C, Zhang J, Wang M, Luo X, Hou Z (2020) Contemporaneous measurement of outer and inner membrane permeability in Gram-negative Bacteria. Bio Protoc 10:3548. https://doi.org/10.21769/bioprotoc.3548
Mroczkowski BS, Huvar A, Lernhardt W, Misono K, Nielson K, Scott B (1994) Secretion of thermostable DNA polymerase using a novel baculovirus vector. J Biol Chem 269:13522–13528
Nelson DE, Ghosh AS, Paulson AL, Young KD (2002) Contribution of membrane-binding and enzymatic domains of penicillin binding protein 5 to maintenance of uniform cellular morphology of Escherichia coli. J Bacteriol 184:3630–3639
Pang C, Liu S, Zhang G, Zhou J, Du G, Li J (2022) Enhancing extracellular production of lipoxygenase in Escherichia coli by signal peptides and autolysis system. Microb Cell Fact 19(1):21
Payne WJ, Eagon RG, Williams AK (1961) Some observations on the physiology of Pseudomonas natriegens nov. spec. Antonie van Leeuwenhoek Journal of Microbiology and Serology 27:121–128
Pembrey RS, Marshall KC, Schneider RP (1999) Cell surface analysis techniques: what do cell preparation protocols do to cell surface properties? Appl Environ Microbiol 65:2877–2894
Pfeifer E, Michniewski S, Gätgens C, Münch E, Müller F, Polen T, Millard A, Blombach B, Frunzke J, Stabb EV (2019) Generation of a prophage-free variant of the fast-growing bacterium Vibrio natriegens. Appl Environ Microbiol 85(17):00853–00819
Pluthero FG (1993) Rapid purification of high-activity taq. DNA polymerase Nucleic Acids Res 21:4850–4851. https://doi.org/10.1093/nar/21.20.4850
Quax WJ (1997) Merits of secretion of heterologous proteins from industrial microorganisms. Folia Microbiol 42:99–103. https://doi.org/10.1007/bf02898715
Schwarz S, Gerlach D, Fan R, Czermak P (2022) GbpA as a secretion and affinity purification tag for an antimicrobial peptide produced in Vibrio natriegens. Electron J Biotechnol 56:75–83. https://doi.org/10.1016/j.ejbt.2022.01.003
Sørensen HP, Mortensen KK (2005) Advanced genetic strategies for recombinant expression in Escherichia coli. J Biotechnol 115:113–128. https://doi.org/10.1016/j.jbiotec.2004.08.004
Suzuki H, Nishimura Y, Hirota Y (1978) On the process of cellular division in Escherichia coli: a series of mutants of E. coli altered in the penicillin-binding proteins. Proc Natl Acad Sci USA 75:664–668. https://doi.org/10.1073/pnas.75.2.664
Varnado CL, Goodwin DC (2004) System for the expression of recombinant hemoproteins in Escherichia coli. Protein Exp Purif 35(1):76–83. https://doi.org/10.1016/j.pep.2003.12.001
Weinstock MT, Hesek ED, Wilson CM, Gibson DG (2016) Vibrio natriegens as a fastgrowing host for molecular biology. Nat Methods 13:849–851. https://doi.org/10.1038/nmeth.3970
Yang J, Moyana T, MacKenzie S, Xia Q, Xiang J (1998) One hundred seventy-fold increase in excretion of an FV fragment-tumor necrosis factor alpha fusion protein (sFV/TNF-alpha) from Escherichia coli caused by the synergistic effects of glycine and triton X-100. Appl Environ Microbiol 64:2869–2874. https://doi.org/10.1128/aem.64.8.2869-2874.1998
Yang H, Hu J, Lu X, Wang F, Shen W, Hu W, Wang L, Chen X, Liu L (2019) Improving extracellular protein production in Escherichia coli by overexpressing D,D-carboxypeptidase to perturb peptidoglycan network synthesis and structure. Appl Microbiol Biotechnol 103:793–806. https://doi.org/10.1007/s00253-018-9510-7
Yoon SH, Kim SK, Kim JF (2010) Secretory production of recombinant proteins in Escherichia coli. Recent Pat Biotechnol 4:23–29. https://doi.org/10.2174/187220810790069550
Yousif SY, Broome-Smith JK, Spratt BG (1985) Lysis of Escherichia coli by β-lactam antibiotics: deletion analysis of the role of penicillin-binding proteins 1A and 1B.J. Gen. Microbiol 131:2839–2845. https://doi.org/10.1099/00221287-131-10-2839
Acknowledgements
We are thankful to Prof. Dr. Julia Frunzke (Forschungszentrum Jülich GmbH, Institute for Bio- and Geosciences) for providing us with a Prophage-free strain of V. natriegens for purposes of this work.
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic. This research was supported by Slovak Research and Development Agency grants (APVV-17-0333, APVV-19-0196, APVV-20-0284, APVV 21-0215, APVV-17-0570) and by the project UpScale of Comenius University Capacities and Competence in Research, Development and Innovation, ITMS: 313021BUZ3, co-financed by the European Regional Development Fund within the Operational Programme Integrated Infrastructure.
Author information
Authors and Affiliations
Contributions
Conceptualization: [Ľubica Kormanová]; Methodology: [Ľubica Kormanová, Zdenko Levarski]; Formal analysis and investigation: [Ľubica Kormanová; Lenka Levarská]; Writing - original draft preparation: [Ľubica Kormanová; Zdenko Levarski; Andrej Minich]; Writing - review and editing: [all authors]; Funding acquisition: [Stanislav Stuchlík]; Resources: [Ján Turňa]; Supervision: [Stanislav Stuchlík]
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kormanová, Ľ., Levarski, Z., Minich, A. et al. Novel expression system based on enhanced permeability of Vibrio natriegens cells induced by D,D- carboxypeptidase overexpression. World J Microbiol Biotechnol 39, 277 (2023). https://doi.org/10.1007/s11274-023-03723-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-023-03723-z