
Abstract
Human metapneumovirus (HMPV) is a major pathogen of acute respiratory tract infections (ARTIs) in children. Whole genome sequence analyses could help understand the evolution and transmission events of this virus. In this study, we sequenced HMPV whole genomes to improve the identification of molecular epidemiology in Beijing, China. Nasopharyngeal aspirates of hospitalized children aged < 14 years old with ARTIs were screened for HMPV infection using qPCR. Fourteen pairs of overlapping primers were used to amplify whole genome sequences of HMPV from positive samples with high viral loads. The epidemiology of HMPV was analysed and 27 HMPV whole genome sequences were obtained. Sequence identity and the positional entropy analyses showed that most regions of HMPV genome are conserved, whereas the G gene contained many variations. Phylogenetic analysis identified 25 HMPV sequences that belonged to a newly defined subtype A2b1; G gene sequences from 24 of these contained a 111-nucleotide duplication. HMPV is an important respiratory pathogen in paediatric patients. The new subtype A2b1 with a 111-nucleotide duplication has become predominate in Beijing, China.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human metapneumovirus (HMPV) is a member of the Pneumoviridae family and was first discovered in 2001 from a patient with a respiratory infection [1]. HMPV mainly causes upper and lower respiratory tract infections in children with mild symptoms. However, HMPV can also occur in concentrated outbreaks among susceptible people, and even cause death in critically ill patients [2]. Notably, in an Iranian case report, three children co-infected with HMPV and severe acute respiratory coronavirus 2 (SARS-COV-2) were found to have died, suggesting that HMPV may directly or indirectly influence susceptibility to and pathogenicity of SARS-CoV-2 [3].
HMPV is a single-stranded RNA virus with a genome of approximately 13 Kb, containing eight genes encoding nine proteins. Based on genomic characteristics, HMPV can be divided into four subtypes, A1, A2, B1, and B2; subtype A2 can be further classified into two additional subtypes A2a and A2b [4]. Although several HMPV subtypes can be prevalent at the same time every year, there is a dominant subtype. Recently, a new genotype of A2 was defined in several studies [5,6,7], and a novel mutant that contained a 180-nucletoide (nt) or 111-nt duplication (dup) in the G gene has been detected since 2015 [8,9,10]. The clinical role and genotype of this new mutant is still unclear, and whole genome sequence monitoring is necessary.
In this study, positive HMPV respiratory infection samples collected from children in Beijing between 2017 and 2019 were Sanger sequenced. A total of 27 HMPV whole genome sequences were obtained and analysed to study the sequence characteristics of HMPV whole genomes in Beijing, China.
Materials and methods
Study population and specimen collection
Nasopharyngeal aspirates (NPAs) were collected from children (aged < 14 years old) with acute respiratory tract infections (ARTIs) at the Beijing Friendship Hospital (China) between April 2017 to March 2019. Clinical characteristics were recorded. All samples were collected in tubes containing viral transport medium and kept at − 80 °C until use.
Detection of HMPV
Total viral nucleic acid was extracted from 200 μL of each clinical specimen using a QIAamp MinElute Kit (Qiagen, Germany). HMPV detection was performed by quantitative real-time polymerase chain reaction (qPCR) assay using a One-step RT-PCR Kit (Ambion, USA). The specific primers (Forward primer: 5'-CATATAAGCATGCTATATTAAAAGAGTCTC-3'; Reverse primer: 5'-CCTATTTCTGCAGCATATTTGTAATCAG-3') and probes (5'-FAM-TGYAATGATGAGGGTGTCACTGCGGTTG-3'-TAMRA) for the HMPV N gene were used as previously described [11]. Samples with a cycle threshold (Ct) < 37 were regarded as positive.
Sequencing
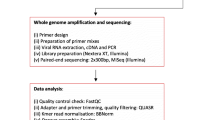
HMPV-positive samples with high copy numbers (Ct < 30) were further used for whole genome sequencing. Viral nucleic acid was reverse transcribed using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, USA) to synthesize cDNA. Whole genomic sequences were amplified by fourteen pairs of overlapping primers that were used as previously described [12]. PCR was performed with Ex-Taq (TaKaRa, China), and amplification products were sequenced by Beijing Tsingke Biological Technology. For fragments that were difficult to amplify, new nested primers were designed for amplification. Obtained sequences were assembled using Sequencher 5.0 software.
Phylogenetic and evolutionary dynamics analysis
Sequence comparisons were made using published GenBank HMPV sequences, and 30 HMPV whole genome sequences and 52 HMPV gene F sequences were chosen for phylogenetic analysis. Neighbor-joining (NJ) trees were constructed using MEGA 7.0 with 1000 bootstrap replicates. The sequence identity and the entropy values of amino acid residues encoded in the whole genome sequences were calculated using BioEdit. Recombination events were detected by Simplot. In addition, 554 G gene sequences of HMPV genotype-A from 1982 to 2019 were retrieved from GenBank database (Supplementary Table 1). Phylogenetic and evolutionary dynamics analyses on HMPV genotype A were performed using the Bayesian Markov chain Monte Carlo (MCMC) methods as implemented in BEAST v1.10.4. Two independent runs were performed for 100 million generations, sampling every 10,000 MCMC step, under a Tamura-Nei (TN93) + Int substitution model and the lognormal relaxed molecular clock. The population dynamics of HMPV genotype-A sequences were estimated by means of Bayesian skyline reconstruction.
Statistical analysis
Data analysis was performed using SPSS 13.0, and the significance of the difference in rates among categorical data was tested by chi-squared and Fisher’s exact tests. Independent-samples t-test was used to analyze continuous variables. Two-tailed P < 0.05 was considered statistical significance.
Results
Epidemiology of HMPV
From April 2017 to March 2019, we collected a total of 2,848 NPAs in the Beijing Friendship hospital (China), and 176 samples (6.18%, 176/2828) were shown to be HMPV positive via qPCR. The male/female ratio of HMPV infection was 1.55 (107:69), which has no significant difference (p = 0.128). The median age of HMPV-infected children was 3 years (IQR: 0–14 years), and the detection rates have no significantly different among different age groups (p = 0.093) (Table1). However, the HMPV prevalence of patients aged ≤ 4 years (139/2026, 6.86%) was significantly higher than that of patients aged 4–14 years (37/822, 4.50%) (p = 0.018). HMPV infection from April 2017 to March 2019 has significant seasonal differences, mainly occurred in spring (42.05%, 74/176) and winter (37.50%, 66/176) (p < 0.001). In contrast, the HMPV detection rate in summer and autumn is 9.66% (17/176) and 10.80% (19/176), respectively.
Clinical characteristics of HMPV infections
All 176 HMPV-infected patients were diagnosed with a lower respiratory tract infection (LRTI). The average length of hospital stay was 6.47 ± 2.45 days, and 61.93% HMPV-infected children were discharged within 7 days. The main clinical symptoms included cough (96.02%, 169/176), fever (≥ 38 °C; 91.48%,161/176), rhinorrhoea (51.14, 90/176), and other common flu-like symptoms and gastrointestinal symptoms. None of the patients developed severe respiratory problems, such as dyspnea. The mean viral load of HMPV-positive samples was 6.7 × 104 copies/μL, and there was no significant difference in viral load between patients aged 0–4 years and patients aged 5–14 years, and HMPV infected patients under 4 years of age were more likely to have respiratory symptoms than patients aged 5–14 years, such as rhinorrhoea (58.99% vs 21.62%, p < 0.001) and nasal obstruction (29.5% vs 16.22%) (Table 2).
Whole genome analysis of HMPV
General PCR was performed on HMPV positive samples with a Ct value < 30, and the products were Sanger sequenced. The fragments were assembled to produce whole genome sequences, and 27 HMPV genomes were obtained. The sequence lengths were between 13,281 and 13,448 nt in length, and the sequences have been submitted to GenBank, the GenBank accession numbers are provided in Supplementary Table 2. The sequence identity among all the analysed sequences was 0.799–0.999 and was 0.801–0.999 among the sequences identified in this study (Table 3). The sequence positional entropy (SPE) was calculated for each residue position of the aligned sequences. Most protein coding regions exhibited low SPE values, whereas the amino acid residues encoded by the G gene had high SPE values (Fig. 1). The Simplot analysis showed no evidence of recombination for any of the sequences in this study. The HMPV F protein is an important viral protein that mediates the membrane fusion reaction, which mainly relies on two functional structural sites (cleavage site: RQSR and integrin αvβ1 binding site: RGD) [13, 14], the SNP analysis of these two functional structure sites showed no mutation among the 27 HMPV F genes.

Sequence positional entropy (SPE) analyses of amino acid residues of the HMPV protein coding region
Phylogenetic analysis of HMPV
A phylogenetic tree of 27 HMPV whole genome sequences was constructed using NJ with the Tamura-Nei model. Based on this, HMPV can be divided into six sub-lineages (A1, A2a, A2b1 A2b2, B1, and B2). Among the 27 sequences, 25 (92.59%) were grouped into the A2b lineage, and the other two sequences were grouped into the B1 lineage (Fig. 2). Phylogenetic analyses of HMPV F gene sequences had the same results as the whole genome phylogenetic analyses. There are three sub-lineages in A2 (A2a, A2b1, and A2b2), and all 25 detected A2 sequences were classed as the A2b1 subtype. The alignment results showed that 96% of obtained A2b1 sequences (24/25) contained a 111-nt-dup in the G gene.

Phylogenetic relationships of 27 HMPV strains. A Neighbor-joining (NJ)-tree based on the whole genome sequences. B NJ-tree based on the F gene sequences. The strains obtained in this study are marked with ●
To understand the demographic history of HMPV A genotype, 554 global HMPV G gene sequences were collected from GenBank, and the Bayesian Maximum Clade Credibility (MCC) trees were defined Fig. 3). The Bayesian skyline population estimate indicated that the population dynamic of the HMPV A genotype peaked between 2000 and 2003, and then plateaued from 2003 to 2009; after 2009, there was a further decline in population size. All G-180-nt-dup and G-111-nt-dup sequences were grouped into the A2b1 subtype. The A2b1 lineage most likely originated around 1992, whereas the A2b2 lineage probably originated from 1995. The time to the most recent common ancestor (tMRCA) of the HMPV genotype A2b1-180-nt-dup sequences and A2b1-111-nt-dup sequences was estimated at approximately 7.283 and 4.768 years, respectively. The A2b1-111-nt-dup sequences were derived from A2b1-180-nt-dup sequences. The evolutionary rate of HMPV genotype-A G gene was approximately 3.654 × 10–3 substitution/site/year (95% highest probability density of 3.1303–4.0357). The sequence identity between A2b1 and A2b2 was 0.615–0.968, and the sequence identity between A2b1-180-nt-dup and A2b1-111-nt-dup was 0.847–0.89.

Demographic history of HMPV G gene. A Population growth inferred via a Bayesian Skyline coalescent tree. The blue lines represent the 95% highest probability density of the effective population size while the yellow line indicates the median of the size and genetic diversity of the population. B Maximum clade credibility tree of the HMPV G gene constructed using BEAST (version 1.10.5) under the skygrid nonparametric coalescent model. The green box indicates A2b1 180-nt-dup strains, and the yellow box indicates A2b1 111-nt-dup strains
Discussion
The serological study showed that HMPV has been globally circulating since 1958 and was identified late in 2001 because of the difficulty encountered in isolating and culturing this virus in vitro [1]. Recently, the novel variants with G-111-nt and G-180-nt dup variants have been detected in countries around the world, including Japan, Spain, Croatia, and China [8,9,10, 15,16,17], highlighting the risks of HMPV epidemics. In this study, we obtained 27 HMPV genome sequences from NPAs of hospitalized children with ARTIs and, using PCR and Sanger sequencing, analyzed the phylogenetics of the HMPV sequences, especially the HMPV A genotype.
HMPV mainly causes respiratory infection in children between the ages of 2 and 5, although reinfection can occur throughout life [18]. HMPV mainly causes upper and/or lower respiratory tract infections while the clinical characteristics are difficult to distinguish from those of influenza-like symptoms. Lower respiratory tract infections due to HMPV can lead to pneumonia, bronchiolitis, and acute asthma exacerbations [19]. In this study, we screened samples from hospitalized children with respiratory infections over two years, and the HMPV infection rate was 6.18% (176/2828). HMPV infection has no gender significant difference. The median age of HMPV-infected children was 36 months, and the HMPV prevalence of patients aged ≤ 4 years was significantly higher than that of patients aged 4–14 years (6.86% vs 4.50%, p = 0.018). The season distribution result showed that HMPV infection mainly occurred in spring (42.05%) and winter (37.50%), which was consistent with prior studies [20, 21] ALL HMPV positive patients were diagnosed with LRTI, main clinical symptoms included cough, fever and rhinorrhoea. HMPV infected patients under 4 years of age were more likely to have respiratory symptoms than patients aged 5–14 years, such as rhinorrhoea (58.99% vs 21.62%, p < 0.001) and nasal obstruction (29.5% vs 16.22%).
The sequence identity and the sequence positional entropy analyses showed that HMPV genome sequences have low genetic diversity, and no recombination event was detected in 27 obtained sequences. The two protein coding regions most susceptible to mutation are the G and SH genes, which is consistent with a previous report [22, 23]. The HMPV F gene has two important functional structural sites (cleavage site: RQSR and integrin αvβ1 binding site: RGD), and no mutation was detected in this study.
THMPV has five sub-genotypes (A1, A2a and A2b, B1, and B2). In recent years, the novel subtypes A2c, A2b1, and A2b2 were proposed based on a short portion of the F gene and limited numbers of HMPV strains [6, 24], and these subtypes have subsequently been identified by a variety of methods. However, Nao, et al. [25] have demonstrated that subtypes A2b2 and A2c are separate descriptions indicating the same subtype, and the detailed evolutionary relationships between these subtypes are still unclear.
In this study, a NJ-Tree was constructed using HMPV whole genome sequences, the result showed that A2b can be divided into 2 clusters, A2b1 and A2b2. In the 27 obtained sequences, 25 (92.59%) grouped into the A2b1 lineage, and the other two sequences grouped into the B1 lineage. Phylogenetic analyses of HMPV F gene sequences showed the same results, and 96% of these sequences contained an 111-nt dup in the G gene, which is consistent with results from Japan and Luohe, China [17, 26]. Although the G protein is unnecessary for HMPV infection [27], it may improve the transmission of the HMPV subgroup A2b strain [26]. The same nucleotide duplication events have also been seen in the G gene of the human respiratory syncytial virus, another member of the Pneumoviridae family, which has become the dominant strain globally [28,29,30]. The MCC tree results showed that the A2b genotype formed two branches as early as 1992, and the A2b1-180-nt dup lineage most likely originated in 2011, which is consistent with a previous study [8]. A2b1-111-nt-dup sequences were derived from A2b1-180-nt dup sequences around 2014. The evolutionary rate of the HMPV A genotype G gene was approximately 3.654 × 10–3 substitution/site/year as shown previously [8]. The demographic population history of HMPV genotype A implied outbreaks in effective population size since 2001; this may have been caused by the discovery of HMPV in 2001, and many studies have focused on the evolution of HMPV since then. However, after 2009, there was a decline in HMPV genotype A population size, this may be due to the improve of medical and health conditions in many places.,.
In conclusion, HMPV is an important virus in paediatric patients, especially in children between 2 and 5 years old. In this study, 27 HMPV whole genome sequences were obtained, and the character of their whole genome sequences were analysed. Most regions of the HMPV genome were conserved, although those of the G gene was the most variable. Phylogenetic analysis showed that 25 obtained HMPV sequences belong to a newly defined subtype A2b1 and that the G gene of 24 sequences contained a 111-nt duplication. The role of the 180-nt-dup and 111-nt-dup remains unclear, which is a reminder for us to continuously monitor this new subtype and investigate the pathogenicity of the new subtype.
Data Availability
Availability of data and materials condensed anonymized data are available from the corresponding author on reasonable request.
References
van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA et al (2001) A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7:719–724. https://doi.org/10.1038/89098
Peña SA, Davis SS, Lu X, Sakthivel SKK, Peret TCT, Rose EB et al (2019) Severe respiratory illness associated with Human metapneumovirus in Nursing Home, New Mexico, USA. Emerg Infect Dis 25:383–384. https://doi.org/10.3201/eid2502.181298
Hashemi SA, Safamanesh S, Ghasemzadeh-Moghaddam H, Ghafouri M, Mohajerzadeh-Heydari MS, Namdar-Ahmadabad H et al (2021) Report of death in children with SARS-CoV-2 and Human metapneumovirus (hMPV) coinfection: Is hMPV the trigger? J Med Virol 93:579–581. https://doi.org/10.1002/jmv.26401
Huck B, Scharf G, Neumann-Haefelin D, Puppe W, Weigl J, Falcone V (2006) Novel Human metapneumovirus sublineage. Emerg Infect Dis 12:147–150. https://doi.org/10.3201/eid1201.050772
Jagušić M, Slović A, Ljubin-Sternak S, Mlinarić-Galinović G, Forčić D (2017) Genetic diversity of Human metapneumovirus in hospitalized children with acute respiratory infections in Croatia. J Med Virol 89:1885–1893. https://doi.org/10.1002/jmv.24884
Nidaira M, Taira K, Hamabata H, Kawaki T, Gushi K, Mahoe Y et al (2012) Molecular epidemiology of Human metapneumovirus from 2009 to 2011 in Okinawa, Japan. Jpn J Infect Dis 65:337–340. https://doi.org/10.7883/yoken.65.337
Chow WZ, Chan YF, Oong XY, Ng LJ, Nor ES, Ng KT et al (2016) Genetic diversity, seasonality and transmission network of Human metapneumovirus: identification of a unique sub-lineage of the fusion and attachment genes. Sci Rep 6:27730. https://doi.org/10.1038/srep27730
Saikusa M, Kawakami C, Nao N, Takeda M, Usuku S, Sasao T et al (2017) 180-Nucleotide duplication in the G Gene of Human metapneumovirus A2b subgroup strains circulating in Yokohama City, Japan, since 2014. Front Microbiol 8:402. https://doi.org/10.3389/fmicb.2017.00402
Piñana M, Vila J, Gimferrer L, Valls M, Andrés C, Codina MG et al (2017) Novel Human metapneumovirus with a 180-nucleotide duplication in the G gene. Future Microbiol 12:565–571. https://doi.org/10.2217/fmb-2016-0211
Saikusa M, Nao N, Kawakami C, Usuku S, Sasao T, Toyozawa T et al (2017) A novel 111-nucleotide duplication in the G gene of Human metapneumovirus. Microbiol Immunol 61:507–512. https://doi.org/10.1111/1348-0421.12543
Maertzdorf J, Wang CK, Brown JB, Quinto JD, Chu M, de Graaf M et al (2004) Real-time reverse transcriptase PCR assay for detection of Human metapneumoviruses from all known genetic lineages. J Clin Microbiol 42:981–986. https://doi.org/10.1128/jcm.42.3.981-986.2004
Wang C, Wei T, Ma F, Wang H, Guo J, Chen A et al (2021) Epidemiology and genotypic diversity of Human metapneumovirus in paediatric patients with acute respiratory infection in Beijing. China Virol J 18:40. https://doi.org/10.1186/s12985-021-01508-0
Schickli JH, Kaur J, Ulbrandt N, Spaete RR, Tang RS (2005) An S101P substitution in the putative cleavage motif of the Human metapneumovirus fusion protein is a major determinant for trypsin-independent growth in vero cells and does not alter tissue tropism in hamsters. J Virol 79:10678–10689. https://doi.org/10.1128/jvi.79.16.10678-10689.2005
Cseke G, Maginnis MS, Cox RG, Tollefson SJ, Podsiad AB, Wright DW et al (2009) Integrin alphavbeta1 promotes infection by Human metapneumovirus. Proc Natl Acad Sci U S A 106:1566–1571. https://doi.org/10.1073/pnas.0801433106
Oketch JW, Kamau E, Otieno GP, Otieno JR, Agoti CN, Nokes DJ (2019) Human metapneumovirus prevalence and patterns of subgroup persistence identified through surveillance of pediatric pneumonia hospital admissions in coastal Kenya, 2007–2016. BMC Infect Dis 19:757. https://doi.org/10.1186/s12879-019-4381-9
Jagusic M, Slovic A, Ivancic-Jelecki J, Ljubin-Sternak S, Vilibić-Čavlek T, Tabain I et al (2019) Molecular epidemiology of human respiratory syncytial virus and Human metapneumovirus in hospitalized children with acute respiratory infections in Croatia 2014–2017. Infect Genet Evol 76:104039. https://doi.org/10.1016/j.meegid.2019.104039
Xie Z, Xu J, Ren Y, Cui A, Wang H, Song J et al (2021) emerging Human metapneumovirus gene duplication variants in patients with severe acute respiratory infection, China, 2017–2019. Emerg Infect Dis 27:275–277. https://doi.org/10.3201/eid2701.201043
Divarathna MVM, Rafeek RAM, Noordeen F (2020) A review on epidemiology and impact of Human metapneumovirus infections in children using TIAB search strategy on PubMed and PubMed Central articles. Rev Med Virol 30:e2090. https://doi.org/10.1002/rmv.2090
Panda S, Mohakud NK, Pena L, Kumar S (2014) Human metapneumovirus: review of an important respiratory pathogen. Int J Infect Dis 25:45–52. https://doi.org/10.1016/j.ijid.2014.03.1394
Xiao NG, Xie ZP, Zhang B, Yuan XH, Song JR, Gao HC et al (2010) Prevalence and clinical and molecular characterization of Human metapneumovirus in children with acute respiratory infection in China. Pediatr Infect Dis J 29:131–134. https://doi.org/10.1097/inf.0b013e3181b56009
Yi L, Zou L, Peng J, Yu J, Song Y, Liang L et al (2019) Epidemiology, evolution and transmission of Human metapneumovirus in Guangzhou China, 2013–2017. Sci Rep 9:14022. https://doi.org/10.1038/s41598-019-50340-8
Piyaratna R, Tollefson SJ, Williams JV (2011) Genomic analysis of four Human metapneumovirus prototypes. Virus Res 160:200–205. https://doi.org/10.1016/j.virusres.2011.06.014
Biacchesi S, Skiadopoulos MH, Boivin G, Hanson CT, Murphy BR, Collins PL et al (2003) Genetic diversity between Human metapneumovirus subgroups. Virology 315:1–9. https://doi.org/10.1016/s0042-6822(03)00528-2
Regev L, Meningher T, Hindiyeh M, Mendelson E, Mandelboim M (2012) Increase Human metapneumovirus mediated morbidity following pandemic influenza infection. PLoS One 7:e34750. https://doi.org/10.1371/journal.pone.0034750
Nao N, Saikusa M, Sato K, Sekizuka T, Usuku S, Tanaka N et al (2020) Recent molecular evolution of Human metapneumovirus (HMPV): subdivision of HMPV A2b strains. Microorganisms. https://doi.org/10.3390/microorganisms8091280
Saikusa M, Nao N, Kawakami C, Usuku S, Tanaka N, Tahara M et al (2019) Predominant detection of the subgroup A2b Human metapneumovirus strain with a 111-nucleotide duplication in the G gene in Yokohama City, Japan in 2018. Jpn J Infect Dis 72:350–352. https://doi.org/10.7883/yoken.JJID.2019.124
Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR et al (2004) Recombinant Human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J Virol 78:12877–12887. https://doi.org/10.1128/jvi.78.23.12877-12887.2004
Trento A, Galiano M, Videla C, Carballal G, García-Barreno B, Melero JA et al (2003) Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 84:3115–3120. https://doi.org/10.1099/vir.0.19357-0
Trento A, Viegas M, Galiano M, Videla C, Carballal G, Mistchenko AS et al (2006) Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J Virol 80:975–984. https://doi.org/10.1128/jvi.80.2.975-984.2006
Duvvuri VR, Granados A, Rosenfeld P, Bahl J, Eshaghi A, Gubbay JB (2015) Genetic diversity and evolutionary insights of respiratory syncytial virus A ON1 genotype: global and local transmission dynamics. Sci Rep 5:14268. https://doi.org/10.1038/srep14268
Acknowledgements
We thank Mike Herbert, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn), for editing the English text of a draft of this manuscript.
Funding
This work was supported by the Research on key technologies supporting the National Core Biological Resource Center, National Key R&D Program of China (2022YFC2602202).
Author information
Authors and Affiliations
Contributions
TW., CW. and LZ. conceived and designed the experiments; CW., FM., JG., AC. and ZX. performed the experiments; CW., YH. and ZX. analyzed the data; TW. and ZX. contributed reagents/materials/analysis tools; TW., CW. and LZ. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The project was approved by the Ethical Committee of National Institute for Viral Disease Control and Prevention, China CDC, and the committee's reference number is IVDC2017-021.
Additional information
Edited by Juergen Richt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11262_2023_2001_MOESM1_ESM.xlsx
Supplementary file1 (XLSX 12 KB)—Supplementary Table 1. The GenBank accession numbers of 554 HMPV genotype-A G gene sequences.
11262_2023_2001_MOESM2_ESM.xlsx
Supplementary file2 (XLSX 23 KB)—Supplementary Table 2. The GenBank accession numbers of 27 HMPV whole genome sequences.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wei, T., Wang, C., Ma, F. et al. Whole genome sequencing and evolution analyses of Human metapneumovirus. Virus Genes 59, 524–531 (2023). https://doi.org/10.1007/s11262-023-02001-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-023-02001-2