
Abstract
In this study, three new mycoviruses were identified co-infecting the apple replant disease (ARD)-associated root endophyte Rugonectria rugulosa. After dsRNA extraction, six viral fragments were visualized. Four fragments belong to a quadrivirus, which has a genome size of 17,166 bp. Each of the fragments of this quadrivirus has a single ORF encoding a protein. Two of these proteins are coat protein subunits, one ORF encodes the RdRp, and one protein has an unknown function. This virus was tentatively named rugonectria rugulosa quadrivirus 1 (RrQV1) as a member of the proposed new species Quadrivirus rugonectria. Another fragment represents the dsRNA intermediate form of a + ssRNA mitovirus with a genome size of 2410 nt. This virus encodes an RdRp and is tentatively called rugonectria rugulosa mitovirus 1 (RrMV1). RrMV1 is suggested as a member of a new species with the proposed name Mitovirus rugonectria. The sixth fragment belongs to the genome of an unclassified dsRNA virus tentatively called rugonectria rugulosa dsRNA virus 1 (RrV1). The monopartite dsRNA genome of RrV1 has a length of 8964 bp and contains two ORFs encoding a structure/gag protein and an RdRp. Full genomic sequences were determined and the genome structure as well as molecular properties are presented. After phylogenetic studies and sequence identity analyses, all three isolates are proposed as new mycoviruses. The results help to improve the understanding of the complexity of the factors involved in ARD and support the interest in mycoviral research. Subsequent analyses need to focus on the impact of mycoviruses on the biology and pathogenicity of ARD-associated fungi. The results of such studies could contribute to the development of mitigation strategies against the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the first proven infection of a fungus with a mycovirus in 1962, these viruses and their effect on the host have increasingly been part of intensive research [1]. To date, viral infections have been reported in all main fungal taxa [2, 3]. The majority of these identified viruses have RNA genomes, mostly appearing to be constituted of dsRNA [4]. DsRNA mycoviruses are assigned to 8 taxonomic families and one genus, whereas those with an ssRNA genome are separated in 12 families, of which 11 families are based on + ssRNA viruses and just one on -ssRNA viruses [5]. Up to now, less is known about DNA mycoviruses. Since the first report of a geminivirus-related DNA mycovirus in 2010, just a few more were detected [6,7,8]. Mycoviral infections are usually persistent, but often seem to not affect the phenotypes of the respective host [5]. However, many mycoviruses are known to have either a hypo- or hypervirulent effect on their host fungi [9,10,11,12,13,14,15]. In the case of hypovirulence, an infection causes a decrease in host pathogenicity. On the contrary case of hypervirulent viral infections, the pathogenic effect of the host fungus is enhanced. In this study, viral infections of an endophytic Rugonectria rugulosa isolate were investigated. The fungus was isolated from apple plant roots (Malus x domestica, Borkh.; M26), suffering from apple replant disease (ARD). ARD is a worldwide problem, occurring in orchards when apple is planted repeatedly. The disease is caused by plant reactions due to changes in their (micro-) biome [16]. When affected, apple orchards can lose about 50% profitability by reduction of yield, tree vigor and a possible delay of 2–3 years, after which the trees begin to bear fruit [17]. The causes for ARD are highly complex and include oomycetes, bacteria, nematodes, fungi, and others [16].
In the search for factors of ARD etiology, Nectriaceae species such as R. rugulosa are found repetitively. By sampling apple roots grown in ARD suffering soils, R. rugulosa was found together with related Nectriaceae species in all soils of three ARD affected sites in Germany [18]. Thus, bioassays have shown that fungi of this family can cause ARD symptoms after isolation and re-inoculation [19].
Since ARD is still not fully understood, all contributing factors, such as mycoviruses, have to be taken into consideration. Understanding a possible hypo- or hypervirulence of the occurring mycoviruses might help to investigate on potential applications in biological control and disease management. Thus, the research on mycoviruses is an important component in the development of strategies to mitigate complex plant diseases such as ARD. However, many phytopathogenic fungi are involved in the etiology of the disease and it can be assumed that many of those fungi also carry unidentified viruses. Therefore, the main objective of this study is to give a first insight into the fungal virome of one of the most recurring fungi involved in the etiology of ARD.
Because of the dominance of dsRNA mycoviruses, dsRNA was extracted from R. rugulosa and sequenced by an Illumina system. Sequences were de novo assembled and completed by RACE (Rapid amplification of cDNA ends), followed by annotation of the molecular features of the individual viruses and phylogenetic analyses. The data and full genomic sequences of the three newly identified mycoviruses, presented in this study, are essential for a deeper understanding of the virus–host relationship and form the basis for further research projects.
Materials and methods
Fungal material
The R. rugulosa isolate, used in this study was isolated from ARD suffering, in vitro propagated M26 roots (Malus x domestica, Borkh.), which were grown for 8 weeks in soil from the ARD affected site Ellerhoop (Chamber of Agriculture Schleswig–Holstein, Germany, 53°42′51.7″N, 9°46′12.5″E) [20]. Before isolation, fine root surfaces were disinfected (70% Ethanol 30 s, followed by 7.5 min 2% NaOCl). Root pieces of 1 cm length were plated on water agar (50 µg mL−1 penicillin, 10 µg mL−1 rifampicin, 25 µg mL−1 pimaricin). The outgrown endophytic fungi were separated and cultivated on 2% malt extract agar (MEA). For nucleic acid extraction, the fungi were propagated in 2% malt extract broth for 2 weeks and mycelium was ground in liquid nitrogen. R. rugulosa was identified by PCR and Sanger sequencing, as described by Crous et al. [21], using primers for the histone H3 gene (CYLH3F: 5ʹ AGGTCCACTGGTGGCAAG 3ʹ; CYLH3R: 5ʹ AGCTGGATGTCCTTGGACTG 3ʹ) [22]. The sequencing was performed at Microsynth Seqlab (Göttingen, Germany). The resulting sequences were analyzed by NCBI BLASTn.
The R. rugulosa isolate investigated in this study was named No4. To test the infectivity of No4 after isolation, M26 plants were re-infected using a soil-free inoculation assay as described by Popp et al., 2019 [19]. The plants showed reduced growth, as well as typical blackening in microscopic analyses of fine roots after 5 weeks.
Extraction of nucleic acids
DsRNA for Illumina sequencing was extracted from 20 g ground fungal material, stored at − 80 °C, based on a modified protocol of Morris and Dodds [23] as described by Lesker et al. [24], apart from using a different cellulose (acid-washed powder for column chromatography [Merck; Darmstadt, Germany; product nr. 22,184]). 20 mL eluate was digested first with 20 U Rnase T1 (Roche; Basel, Switzerland) and then with 40 U DNAse I (Roche; Basel, Switzerland) at 37 °C for 30 min each. DsRNA extracts were centrifuged and suspended in 25 µL Tris (5 mM). Subsequently, 20 µL extract was checked with 5 µL of GelRed® (Biotium; Fremont, CA, USA) dye in 1.5% agarose gel electrophoreses. For virus detection by RT-PCR and RNA end determination, a simpler protocol for whole nucleic acid extraction was used, following the protocol of Menzel et al. [25].
Illumina sequencing
A Nextera XT Library Preparation Kit was used to prepare an Illumina library from double-stranded cDNA, obtained by cDNA synthesis of the dsRNA extract and second-strand synthesis with random octamer primers. The library was sequenced at the Leibniz-Institute DSMZ on a NextSeq instrument as paired-end reads (2 × 151 bp). The raw reads were trimmed and de novo assembled with Geneious v. R11.1 software (Biomatters; Auckland, New Zealand) using an in-house established workflow, followed by local BLASTn and BLASTp alignments of the assembled contigs against a custom database of NCBI nuclear-core reference sequences. The identified mycovirus contigs were ordered and trimmed according to reference sequences to determine the nearly complete genome sequences. The sequence information was used to design primers for virus detection by RT-PCR and determination of the extreme terminal ends of the genomes by RACE.
Virus detection with RT-PCR
RT-PCR protocols were adapted for the detection of each of the genomic viral segments. For cDNA synthesis, 4 µL dsRNA extract, 1 µL cDNA primer [10 µM (Table 1); salt-free; Eurofins Genomics; Ebersberg, Germany] and 5 µL A.bidest were mixed and heated up to 95 °C for three minutes to separate the dsRNA strands. 50 U Maxima H Minus Reverse Transcriptase (Thermo Fisher Scientific™; Waltham, MA, USA), 20 U RiboLock RNase Inhibitor (Thermo Fisher Scientific™; Waltham, MA, USA), 1 µL dNTPs (10 mM each; Thermo Fisher Scientific™; Waltham, MA, USA) and 4 µL 5X RT-buffer (250 mM Tris–HCl (pH 8.3), 250 mM KCl, 20 mM MgCl2, 50 mM DTT; Thermo Fisher Scientific™; Waltham, MA, USA) were added subsequently and adjusted to 20 µL with A.bidest. The cDNA synthesis started with 60 min at 50 °C, followed by 15 min at 55 °C, 15 min at 60 °C, and 5 min at 85 °C. PCR was performed with 5 µL 2 × Phusion Flash High-Fidelity PCR Master Mix (Thermo Fisher Scientific™; Waltham, MA, USA), 1 µL of each specific primer [forward and reverse, 10 µM each, salt-free; Eurofins Genomics; Ebersberg, Germany (Table 1)], 2 µL cDNA and 1 µL A.bidest. PCR was performed with an initial denaturation of 15 s at 98 °C, followed by 34 cycles (denaturation: 98 °C, 5 s; annealing: primer TA, 5 s; elongation: 72 °C, 15 s / 1000 bp amplicon), and a final elongation of 300 s at 72 °C. Amplicons were visualized by 1.0% agarose electrophoresis and sent to Microsynth Seqlab (Göttingen, Germany) for Sanger sequencing [22].
RNA end determination
The ends of dsRNAs were determined by RACE with an adapted protocol, based on the method described by Frohman et al. [26]. 3ʹ-ends of both, dsRNA sense and antisense strands were analyzed. Reverse transcription followed the described protocol for virus detection, with different cDNA primers (10 µM each, salt-free; Eurofins Genomics; Ebersberg, Germany; Table 2). For tailing 3 µL cDNA was mixed with 20 U Terminal Deoxynucleotidyl Transferase (TdT; Thermo Fisher Scientific™; Waltham, MA, USA), 4 µL 5 × TdT Reaction Buffer (500 mM potassium cacodylate (pH 7.2), 10 mM CoCl2, 1 mM DTT; Thermo Fisher Scientific™; Waltham, MA, USA), 1 µL of either dATP, dCTP, dGTP, or dTTP (100 mM, Thermo Fisher Scientific™; Waltham, MA, USA) and 11 µL A.bidest. The mixture was incubated for 30 min at 37 °C followed by 10 min at 70 °C. For each RNA end, at least two different tails were used in different reactions. The subsequent PCR was performed as the one for virus detection with a different primer set (poly-n primer; nested primer; 10 µM each, salt-free; Eurofins Genomics; Ebersberg, Germany; Table 2).
Phylogenetic analyses
Phylogenetic analyses were performed with several mycoviruses of the families Totiviridae, Quadriviridae, Chrysoviridae, Mitoviridae, and ten unclassified dsRNA Riboviria viruses. Before constructing a phylogenetic tree, amino acid sequences of the RNA-dependent RNA polymerase of all viruses were aligned, using the MUSCLE algorithm in MEGA X [27, 28]. Parameters were set to default (gap opening: − 2.9, gap extension: 0). After the initial alignment, highly conserved sequences were selected, referring to the segment A(679)–E(1066) of NC_016760 [29]. The final alignment was performed with the set of chosen segments and default parameters, using the MUSCLE algorithm again. A maximum-likelihood tree was calculated, using the bootstrap method with 1000 replications and the Le_Gascuel_2008 substitution model with discrete Gamma distribution (LG + G) [30]. The number of discrete gamma categories was set to 5 and for the data subset, all sites were used. Pairwise alignments of all segments of the families Quadriviridae, Mitoviridae, and the unclassified dsRNA viruses were done to calculate sequence identities by using the EMBOSS/Needle tool [31].
UTR alignment, secondary structures, and motifs
Conserved UTR-sequences of the proposed quadrivirus RrQV1 were analyzed by alignments of the four 5ʹ- and 3ʹ-ends and presented with the GeneDoc Software (National Resource for Biomedical Supercomputing, Pittsburgh, PA, USA). Secondary structure predictions of the proposed mitovirus RrMV1 were computed with the RNAfold WebServer (Institute for Theoretical Chemistry, University of Vienna) [32, 33]. Conserved motifs within the genomic RNAs were identified by using the NCBI Conserved Domain Search tool with default options (database: CDD v3.19–—58,235 PSSMs, expected value threshold: 0.01) [34].
Results and discussion
Identification of new mycoviruses
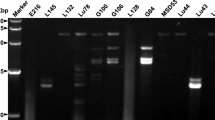
After dsRNA extraction of R. rugulosa, agarose gel electrophoresis enabled the visualization of six fragments (Fig. 1). Illumina sequencing and de novo assembly led to six consensus sequences, which were assigned to different virus families by BLAST. Accordingly, four fragments represent the different segments of a quadrivirus. One fragment belongs to a member of the family Mitoviridae and another one to an unclassified dsRNA virus. Each of the six RNAs could be detected in R. rugulosa by RT-PCR. End determination by 5ʹ- and 3ʹ-RACE led to full genomic RNAs. The four segments of genomic RNAs of the quadrivirus—with the proposed name rugonectria rugulosa quadrivirus 1 (RrQV1)—have sizes of 4897 bp, 4312 bp, 4153 bp, and 3804 bp and each RNA encodes a single open reading frame (ORF). The mitovirus with the suggested name rugonectria rugulosa mitovirus 1 (RrMV1) consists of one RNA with a length of 2410 nt and one ORF. The unassigned virus with the proposed name rugonectria rugulosa dsRNA virus 1 (RrV1) has one genomic RNA, 8964 bp in length, which encodes two ORFs.

1.5% Agarose gel electrophoresis of dsRNA, isolated from Rugonectria rugulosa. Fragments correspond for rugonectria rugulosa quadrivirus 1 (RrQV1), rugonectria rugulosa mitovirus 1 (RrMV1), and rugonectria rugulosa dsRNA virus 1 (RrV1). Nucleic acid stained with GelRed®. Lane 1: dsRNA, Lane 2: PstI digested λ phage DNA. A Original gel, B Detailed view of dsRNA fragments
Subsequent comparison of genome sizes determined from sequence analyses with sizes from the agarose gel of dsRNA extractions reveals that the fragments in the gel, compared to the λ-phage DNA ladder, were all higher than expected. Such a shift can occur when using GelRed® as dye for nucleic acids in agarose gels. Although it is relatively non-toxic, it leads to artifacts in agarose gel electrophoreses. The more nucleic acids are present, the stronger these artifacts become and the more difficult a size determination of the fragments [35]. Additionally, dsRNA generally migrates more slowly in agarose gels than DNA because of a lower overall charge density of RNA, which results from condensation and the development of secondary structures [36, 37]. Condensation is comparatively more likely in RNA because the phosphate groups are more closely adjacent than in DNA [38]. Compared to the other viruses, the fragment of the mitovirus is fainter visible in the gel. Since mitoviruses are + ssRNA viruses, it must be assumed that this fragment is not the genomic but its dsRNA intermediate, which occurs in all + ssRNA viruses [39].
Genome organization
The genome of RrQV1 consists of 17,166 bp, split into four ORFs on four dsRNA segments. 5ʹ-untranslated regions (UTRs) are 49 bp to 62 bp long and 3ʹ-UTRs range from 37 to 380 bp. With sizes ranging from 3804 to 4897 bp, the genomic RNAs meet the requirements for segments of quadriviruses, which should range from 3.5 to 5 kbp [29]. Proteins encoded by the ORFs comprise 1591 amino acids (aa, p1), 1384 aa (p2), 1351 aa (p3), and 1120 aa (p4). These proteins have a calculated molecular mass of 175.5 kDa (p1), 150.9 kDa (p2), 148.9 kDa (p3), and 119.5 kDa (p4), respectively. Protein p1 has a so far unknown function, whereas p2 and p4 are coat protein (cp) subunits. P3 is the RNA-dependent RNA polymerase (RdRp) and has a conserved RT-like domain at aa651–aa1112. Upstream of the respective ORFs within the 5ʹ-UTRs of RrQV1, (CAA)n repeats are localized, which may serve as translational enhancers [29, 40]. The first 11 nucleotides at the 5ʹ-end and the last 16 nucleotides at the 3ʹ-end of RrQV1 are highly conserved (5ʹ YACGAAWAAAC…AUUAGCAAUGYGCGCV 3ʹ). This holds true for all four segments, with exception of the 3ʹ-end of RNA 3, which lacks the last two nucleotides. An alignment of the first and last 30 nucleotides of the RrQV1 segments is shown in Fig. 2.

Alignment of A 5ʹ-UTRs and B 3ʹ-UTRs of RrQV1. Numbers next to the sequences represent the nucleotide position, mapped with GeneDoc 2.7. Black boxes are conserved domains. Boxes in gray have one dissimilar nucleotide in maximum
The genome of the mitovirus RrMV1 is monopartite with a 2410 nt + ssRNA. One single ORF is flanked by a 197 nt 5ʹ-UTR and a 68 nt 3ʹ-UTR. It encodes an 83.9 kDa RdRp with a length of 714 aa. A conserved mitovirus-RNA-Polymerase domain was found at position aa170-aa633. Modeling the structure of the terminal sections of the + ssRNA of RrMV1 resulted in three stem-loop structures at the 5ʹ-end with a negative energy change of − 34.91 kcal mol−1 and two stem-loops at the 3ʹ-end with − 29.95 kcal mol−1. The plain structure predictions are shown in Fig. 3. This formation of secondary structures at the ends of the genome is characteristic in mitoviruses and presumably serves to protect the naked genomic RNA from enzymatic degradation in the host cells [41, 42].

The unclassified virus RrV1 has a monopartite 8964 bp dsRNA genome. A 794 bp 5ʹ-UTR and a 77 bp 3ʹ-UTR surround two ORFs with lengths of 3810 nt and 4086 nt. The translated proteins are a proposed 141.0 kDa structural/gag protein (p1) with a length of 1269 aa and a 151.1 kDa RdRp with a length of 1361 aa. A scaled genome map of the three viruses is shown in Fig. 4.

Scaled genome maps. Green: rugonectria rugulosa quadrivirus 1 (RrQV1); blue: rugonectria rugulosa mitovirus 1 (RrMV1); light gray: rugonectria rugulosa dsRNA virus 1 (RrV1). Digits indicate nucleotide positions of 5ʹ-ends, ORFs, and 3ʹ-ends. Boxes represent ORFs with their function (encoding protein) in brackets. Dark gray boxes: Conserved RdRp domains. Lines represent genomic RNAs. Single line: ssRNA, double lines: dsRNA. Scale bars indicate 1000 nt, 500 nt, and 2000 nt, related to the respective virus
Phylogenetic analyses
For the taxonomic studies, the viruses with the highest BLAST scores to RrQV1, RrMV1, and RrV1 were selected. In addition, representatives of the families Chrysoviridae and Totiviridae were included in the calculations as further mycoviral entities. After RdRp translation and alignment, the most conserved region, represented by amino acids 679–1066 of NC_016760 was selected for each virus, as described by Chiba et al. [29]. This set of reduced-length sequences was used for the further alignment, based on which a phylogenetic tree was created in MEGA X [28]. Based on the tree, shown in Fig. 5 it can be stated that all three introduced viruses belong to their proposed family. RrQV1 clusters together with all other Quadriviridae and has the closest relation to OK077752 in 99% of 1000 bootstraps. The placement of RrQV1 in this cluster reinforces the assignment of this virus to the family. Moreover, the taxonomic proximity of Chrysoviridae, Totiviridae, and Quadriviridae has been shown previously and can be confirmed here [29, 43]. The mitovirus RrMV1 has the closest relation to NC_004052 and a subtree of NC_030862 and NC_012585. It is assigned to the family Mitoviridae, which form their own subtree with two clusters in 100% of the bootstraps. Since these + ssRNA viruses are much simpler in structure than the listed dsRNA viruses and use the genetic code of mold fungi, they were expected to form a distinct group and have a higher taxonomic distance from the other viruses [44, 45]. For this reason, no further outgrouper was necessary for the construction of the phylogenetic tree. According to that tree, RrV1 is assigned to the group of unclassified dsRNA Riboviria. In 96% of the bootstraps, it is listed together with JN671443, JN671444, and NC_033415 to a subtree of this group. The other subtree among the unclassified viruses summarizes mainly Fusagravirus species. RrV1 is therefore not included in the cluster, even if it could be considered as a possible family of its own in the future [46].

Phylogenetic tree, based on the alignment of conserved amino acids (aa) of RNA-dependent RNA polymerases (RdRp), referring to aa 679–1066 of the quadrivirus reference NC_016760. Alignment was performed with the MUSCLE algorithm [27]. The tree was constructed with the Maximum Likelihood method and 1000 bootstraps, using the program MEGA X [28]. The substitution model Le_Gascuel_2008 was used with discrete Gamma distribution (LG + G) [30]. Virus families are provided with colored dots and annotation of the family name. The scale bar is representing the substitutions per site. Numbers, next to the branches are indicating the percentage of trees, bootstrapped in the shown manner. Viruses are annotated with their GenBank Accession Number and virus name
Sequence identities and species demarcation
To determine the sequence identities of the new viruses in relation to the others in the phylogenetic tree, pairwise sequence alignments of the gRNA, RdRp-ORFs, and RdRp amino acid sequences were made. Results of analyses of the RdRp encoding Segment 3 of the quadrivirus RrQV1 with other members of the family led to a high similarity with thelonectria quadrivirus 1 (TQV1), with a protein-sequence identity of 94%. The lowest identity was found in the aa-sequence with RnQV1, isolates W1075 and W726 (30.1%). Table 3 gives an overview of all results, comparing segment 3 of the Quadriviridae. Since the identity of RrQV1 Segment 3 was comparatively high with TQV1, the remaining three segments were analyzed as well and summarized in Table 3. RnQV1 W726 was not included here, because of missing sequence data on segments 1, 2, and 4. As for segment 3, TQV1 has the highest similarities with RrQV1 in the other segments. The protein-sequence identity of those viruses varies from 69.9 to 82.8%. The lowest score was obtained with RnQV1 W1118, segment 1 (18.9%). For the determination of the species, there is no official demarcation value for the family of Quadriviridae [29]. The criteria in other virus families are very diverse and are therefore not suitable for transmission and application in this family. To date, only three different members of the family Quadriviridae are known. All of these viruses have been identified in different hosts. With RrQV1, for the first time, an infection within the genus Rugonectria could be detected. Therefore, we recommend RrQV1 as a member of a new species, tentatively named Quadrivirus rugonectria within the family Quadriviridae.
Identities of the mitovirus RrMV1 with other species vary from 22% (aa-sequence, CpMV1) to 55.3% (ORF, BcMV-2). The highest similarity of the RrMV1 aa-sequence (38.4%) was found with CeMV (NC_012585). Table 4 sums up the gRNA-, ORF- and protein sequencesʹ lengths and identities of all analyzed Mitoviridae members. Following the change in taxa, mitoviruses are no longer assigned to the Narnaviridae as of 2019 but have their own family with the Mitoviridae [47, 48]. There is no species demarcation for the Mitoviridae yet. However, since RrMV1 is below 50% sequence identity in protein sequences compared to all other mitoviruses considered and this was the species demarcation value within the Narnaviridae, we propose it as member of a new species, tentatively named Mitovirus rugonectria within the family Mitoviridae [49].
When comparing the RdRp aa-sequences of the unclassified RrV1 with other unclassified dsRNA Riboviria viruses, identities range from 29.3% (MpDSRV2) to 32.9% (FvV1, MpFV3). Similarities of gRNAs and ORFs are between 42.5% and 48.1%. In Table 5, all calculated sequence identities of RrV1 with other viruses are listed. Since all viruses similar in BLAST are unclassified, RrV1 should be assigned to the unclassified dsRNA Riboviria as well. For unclassified viruses, there are no guidelines for species demarcation. At less than 40%, the amino acid sequence identities of RdRp are also low compared to the other unclassified viruses. Therefore, RrV1 is proposed as a new virus. However, RrV1 cannot be assigned to a (new) species according to the ICTV guideline on naming viruses and virus species, since a new species must be assigned to a genus for binomial naming [50].
Conclusion
For the first time, three new viruses were identified using dsRNA extraction and Illumina sequencing from an endophytic Rugonectria rugulosa isolate associated with ARD. Full-length genomic RNAs were used to tease out the particular features of each virus. By phylogenetic analysis, rugonectria rugulosa quadrivirus 1, as member of the suggested species Quadrivirus rugonectria and rugonectria rugulosa mitovirus 1, as member of the proposed species Mitovirus rugonectria could be assigned to a taxonomic family. Together with rugonectria rugulosa dsRNA virus 1, all three viruses can be proposed as new viruses after analysis of sequence identities with related species. Future experiments need to clarify whether infections with these viruses have a hypo- or hypervirulent effect on the host. Although some mycoviral infections do not appear to have any effect on host infection behavior, there are reports of such virulence increasing and decreasing effects of some mycoviruses [9,10,11,12,13,14,15]. If any of these cases occur, the viruses could serve as a control target or biocontrol agent in the containment of ARD. Building on this, the effect of infection on the etiology of ARD needs to be elaborated. Thus, the presented results can contribute to the growing field of mycovirology and the diversity and spread of these viruses in plant root-associated fungi.
References
Hollings M (1962) Viruses associated with a die-back disease of cultivated mushroom. Nature 196:962–965. https://doi.org/10.1038/196962a0
Ghabrial SA, Castón JR, Jiang D et al (2015) 50-plus years of fungal viruses. Virology 479–480:356–368. https://doi.org/10.1016/j.virol.2015.02.034
Pearson MN, Beever RE, Boine B et al (2009) Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol Plant Pathol 10:115–128. https://doi.org/10.1111/j.1364-3703.2008.00503.x
King AMQ, Adams MJ, Carstens EB et al (eds) (2012) Virus taxonomy: classification and nomenclature of viruses. Ninth report of the international committee on taxonomy of viruses. Elsevier Academic Press, London
Kotta-Loizou I (2021) Mycoviruses and their role in fungal pathogenesis. Curr Opin Microbiol 63:10–18. https://doi.org/10.1016/j.mib.2021.05.007
Khalifa ME, MacDiarmid RM (2021) A mechanically transmitted DNA mycovirus is targeted by the defence machinery of its host Botrytis cinerea. Viruses. https://doi.org/10.3390/v13071315
Li P, Wang S, Zhang L et al (2020) A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci Adv. https://doi.org/10.1126/sciadv.aay9634
Yu X, Li B, Fu Y et al (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Natl Acad Sci USA 107:8387–8392. https://doi.org/10.1073/pnas.0913535107
Olivé M, Campo S (2021) The dsRNA mycovirus ChNRV1 causes mild hypervirulence in the fungal phytopathogen Colletotrichum higginsianum. Arch Microbiol 203:241–249. https://doi.org/10.1007/s00203-020-02030-7
Özkan S, Coutts RHA (2015) Aspergillus fumigatus mycovirus causes mild hypervirulent effect on pathogenicity when tested on Galleria mellonella. Fungal Genet Biol 76:20–26. https://doi.org/10.1016/j.fgb.2015.01.003
Nuss DL (2005) Hypovirulence: mycoviruses at the fungal-plant interface. Nat Rev Microbiol 3:632–642. https://doi.org/10.1038/nrmicro1206
Deng F, Xu R, Boland GJ (2003) Hypovirulence-associated double-stranded RNA from Sclerotinia homoeocarpa is conspecific with ophiostoma novo-ulmi mitovirus 3a-Ld. Phytopathology 93:1407–1414. https://doi.org/10.1094/PHYTO.2003.93.11.1407
Polashock JJ, Hillman BI (1994) A small mitochondrial double-stranded (ds) RNA element associated with a hypovirulent strain of the chestnut blight fungus and ancestrally related to yeast cytoplasmic T and W dsRNAs. Proc Natl Acad Sci USA 91:8680–8684. https://doi.org/10.1073/pnas.91.18.8680
Shapira R, Choi GH, Nuss DL (1991) Virus-like genetic organization and expression strategy for a double-stranded RNA genetic element associated with biological control of chestnut blight. EMBO J 10:731–739. https://doi.org/10.1002/j.1460-2075.1991.tb08004.x
Chu Y-M, Jeon J-J, Yea S-J et al (2002) Double-stranded RNA mycovirus from Fusarium graminearum. Appl Environ Microbiol 68:2529–2534. https://doi.org/10.1128/AEM.68.5.2529-2534.2002
Winkelmann T, Smalla K, Amelung W et al (2019) Apple replant disease: causes and mitigation strategies. Curr Issues Mol Biol 30:89–106. https://doi.org/10.21775/cimb.030.089
van Schoor L, Denman S, Cook NC (2009) Characterisation of apple replant disease under South African conditions and potential biological management strategies. Sci Hortic 119:153–162. https://doi.org/10.1016/j.scienta.2008.07.032
Popp C, Wamhoff D, Winkelmann T et al (2020) Molecular identification of nectriaceae in infections of apple replant disease affected roots collected by harris uni-core punching or laser microdissection. J Plant Dis Prot 127:571–582. https://doi.org/10.1007/s41348-020-00333-x
Popp C, Grunewaldt-Stöcker G, Maiss E (2019) A soil-free method for assessing pathogenicity of fungal isolates from apple roots. J Plant Dis Prot 126:329–341. https://doi.org/10.1007/s41348-019-00236-6
Mahnkopp F, Simon M, Lehndorff E et al (2018) Induction and diagnosis of apple replant disease (ARD): a matter of heterogeneous soil properties? Sci Hortic 241:167–177. https://doi.org/10.1016/j.scienta.2018.06.076
Crous PW, Groenewald JZ, Risède J-M et al (2004) Calonectria species and their cylindrocladium anamorphs: species with sphaeropedunculate vesicles. Stud Mycol 50:415–430
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467. https://doi.org/10.1073/pnas.74.12.5463
Morris TJ, Dodds JA (1979) Isolation and analysis of double-stranded RNA from virus-infected plant and fungal tissue. Phytopathology 69:854. https://doi.org/10.1094/phyto-69-854
Lesker T, Rabenstein F, Maiss E (2013) Molecular characterization of five betacryptoviruses infecting four clover species and dill. Arch Virol 158:1943–1952. https://doi.org/10.1007/s00705-013-1691-x
Menzel W, Jelkmann W, Maiss E (2002) Detection of four apple viruses by multiplex RT-PCR assays with coamplification of plant mRNA as internal control. J Virol Methods 99:81–92. https://doi.org/10.1016/S0166-0934(01)00381-0
Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA 85:8998–9002. https://doi.org/10.1073/pnas.85.23.8998
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Chiba S, Castón JR, Ghabrial SA et al (2018) ICTV virus taxonomy profile: Quadriviridae. J Gen Virol 99:1480–1481. https://doi.org/10.1099/jgv.0.001152
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. https://doi.org/10.1093/molbev/msn067
Madeira F, Park YM, Lee J et al (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47:W636–W641. https://doi.org/10.1093/nar/gkz268
Lorenz R, Bernhart SH, Siederdissen HZ, C, et al (2011) ViennaRNA package 2.0. Algorithm Mol Biol 6:26. https://doi.org/10.1186/1748-7188-6-26
Gruber AR, Lorenz R, Bernhart SH et al (2008) The Vienna RNA websuite. Nucleic Acids Res 36:W70–W74. https://doi.org/10.1093/nar/gkn188
Lu S, Wang J, Chitsaz F et al (2020) CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268. https://doi.org/10.1093/nar/gkz991
Huang Q, Baum L, Fu W-L (2010) Simple and practical staining of DNA with gelred in agarose gel electrophoresis. Clin Lab 56:149–152
Gast FU, Hagerman PJ (1991) Electrophoretic and hydrodynamic properties of duplex ribonucleic acid molecules transcribed in vitro: evidence that A-tracts do not generate curvature in RNA. Biochemistry 30:4268–4277. https://doi.org/10.1021/bi00231a024
Bhattacharyya A, Murchie AIH, Lilley DMJ (1990) RNA bulges and the helical periodicity of double-stranded RNA. Nature 343:484–487. https://doi.org/10.1038/343484a0
Manning GS (1978) The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q Rev Biophys 11:179–246. https://doi.org/10.1017/s0033583500002031
Kumar M, Carmichael GG (1998) Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol Mol Biol Rev 62:1415–1434. https://doi.org/10.1128/MMBR.62.4.1415-1434.1998
Ghabrial SA, Castón JR, Coutts RHA et al (2018) ICTV virus taxonomy profile: Chrysoviridae. J Gen Virol 99:19–20. https://doi.org/10.1099/jgv.0.000994
Khalifa ME, Pearson MN (2013) Molecular characterization of three mitoviruses co-infecting a hypovirulent isolate of sclerotinia sclerotiorum fungus. Virology 441:22–30. https://doi.org/10.1016/j.virol.2013.03.002
Hong Y, Dover SL, Cole TE et al (1999) Multiple mitochondrial viruses in an isolate of the dutch elm disease fungus ophiostoma novo-ulmi. Virology 258:118–127. https://doi.org/10.1006/viro.1999.9691
Kotta-Loizou I, Castón JR, Coutts RHA et al (2020) ICTV virus taxonomy profile: Chrysoviridae. J Gen Virol 101:143–144. https://doi.org/10.1099/jgv.0.001383
Cole TE, Hong Y, Brasier CM et al (2000) Detection of an RNA-dependent RNA polymerase in mitochondria from a mitovirus-infected isolate of the dutch elm disease fungus, Ophiostoma novo-ulmi. Virology 268:239–243. https://doi.org/10.1006/viro.1999.0097
Pritchard AE, Sable CL, Venuti SE et al (1990) Analysis of NADH dehydrogenase proteins, ATPase subunit 9, cytochrome b, and ribosomal protein L14 encoded in the mitochondrial DNA of paramecium. Nucleic Acids Res 18:163–171. https://doi.org/10.1093/nar/18.1.163
Wang L, Zhang J, Zhang H et al (2016) Two novel relative double-stranded RNA Mycoviruses infecting Fusarium poae strain SX63. Int J Mol Sci. https://doi.org/10.3390/ijms17050641
Walker PJ, Siddell SG, Lefkowitz EJ et al (2020) Changes to virus taxonomy and the statutes ratified by the international committee on taxonomy of viruses. Arch Virol 165:2737–2748. https://doi.org/10.1007/s00705-020-04752-x
Lefkowitz EJ, Davison AJ, Siddell S et al (2018) Virus taxonomy: classification and nomenclature of viruses: online report of the international committee on taxonomy of viruses. Nucleic Acids Res 46(D1):D708–D717
Hillman BI, Esteban R (2012) Family Narnaviridae. In: King AMQ, Adams MJ, Carstens EB et al (eds) Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, pp 1055–1060
Zerbini FM, Siddell SG, Mushegian AR et al (2022) Differentiating between viruses and virus species by writing their names correctly. Arch Virol 167:1231–1234. https://doi.org/10.1007/s00705-021-05323-4
Acknowledgements
This work was funded by the Federal Ministry of Education and Research and is part of the ORDIAmur network (FKZ: 031B0025A), which is allocated to the BONARES program.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Research involving human participants and/or animals
The authors declare that no human and/or animal material, data, or cell lines were involved in this study.
Informed consent
The authors declare that no individual rights were infringed during this study. All involved individuals have been informed and gave their consent to publication.
Additional information
Edited by Seung-Kook Choi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pielhop, T.P., Popp, C., Knierim, D. et al. Three new mycoviruses identified in the apple replant disease (ARD)-associated fungus Rugonectria rugulosa. Virus Genes 58, 423–435 (2022). https://doi.org/10.1007/s11262-022-01924-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-022-01924-6