
Abstract
A new emerging flavivirus caused severe egg-drop in poultry and spread quickly across most duck-producing regions of China in 2010. Complete genome sequencing indicated that the virus genome is 10,989 nucleotides in length and possesses typical flavivirus genome organization, 5′ untranslated region (UTR)-Cv-Ci-prM-M-E-NS1-NS2A-NS2B-NS3-NS4A-2K-NS4B-NS5-3′-UTR. The long open reading frame (ORF) encodes 3,425 amino acids (95–10,372 nt). The 94-nucleotide 5′-UTR is of intermediate size and the 617-nucleotide 3′-UTR is quite long relative to those of other flaviviruses. The polyprotein cleavage sites, potential glycosylation sites, distribution of cysteine residues, and 3′-UTR secondary structure were characterized. Phylogenetic analysis of the polyprotein sequences indicates that the HN isolate is closely related to Tembusu viruses of the Ntaya virus group.
Similar content being viewed by others

Introduction
Flaviviruses are arthropod-transmitted viruses that belong to the Flaviviridae family. The flavivirus genome consists of single-stranded, positive-sense RNA, approximately 10.5 kb in length, encoding three structural and seven non-structural proteins in one open reading frame (ORF) [1]. The genome lacks a poly-A segment at the 3′-end. Virions consist of capsid proteins (C), envelope proteins (E), small non-glycosylated proteins (M), and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The E protein, which is glycosylated in most flaviviruses, is located on the virion surface and plays an important role in virus receptor-binding, entry, and fusion.
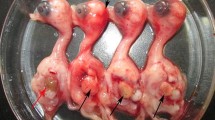
In 2010, an outbreak of unknown infectious severe egg-drop disease spread across most poultry farming regions in China [2–4]. The pathogen caused high morbidity, up to 100 %, and less than 5 % mortality. Egg production rates dropped drastically to 10 % within 5 days. Postmortem examinations showed severe ovarian hemorrhage, ovaritis, and regression. The disease has caused a serious economic loss because of complete elimination of reproduction for some farms.
Here, virus from a sick Muscovy duck from this outbreak was isolated and designated HN strain. To better understand the properties of this causative pathogen, to expand the available flavivirus sequences in public databases, and to establish phylogenetic relationships between the duck flavivirus and others, the full genomic sequence of the HN strain was determined. Features of the HN strain genome, polyprotein cleavage sites, 3′-UTR secondary structure, and 3′-sequence analysis were also characterized.
Materials and methods
Virus propagation
The duck flavivirus HN strain was isolated from the liver of duck with egg-drop symptoms in China in 2010. Briefly, the liver was homogenized in PBS and then centrifuged at 1,000×g for 30 min at 4 °C. The supernatant was passed through a 0.2 μm filter (Whatman, Fisher Scientific, Norcross, GA) and inoculated into the allantoic cavities of 12-day-old embryonated duck eggs. Embryonic viability was monitored daily for 6 days. Virus used in this was passaged three times.
RT-PCR and sequencing
Viral RNA was extracted from virus stock with Trizol reagent (Invitrogen, Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was prepared in accordance with the M-MLV (Invitrogen, Life Technologies, Carlsbad, CA) manufacturer’s instructions. We conducted PCR in a T-Gradient Thermoblock. Samples were subjected to an incubation at 94 °C for 5 min; followed by 30 cycles total of 1-min denaturation at 94 °C, 1-min annealing at 53 °C, and 5-min extension at 65 °C; and a final extension for 10 min at 72 °C. The primers were designed based on the conserved regions of flavivirus genomes; primer sequences will be provided upon request. The 5′- and 3′-ends of the genomes were amplified using 5′-RACE and 3′-RACE kits (Invitrogen, Carlsbad, CA), respectively. For the 5′-RACE, viral RNA was reverse transcribed and the resulting cDNA was incubated in C-tailing buffer. A poly-G primer was used for PCR amplification according to the PCR protocol in the kit. For the 3′-RACE, viral RNA was incubated with poly-A buffer for poly-A tailing of the 3′-terminus. Poly-A-tailed RNA was then amplified by RT-PCR using a forward primer and a reverse primer supplied in the kit. Amplified products were purified and ligated into a pMD18-T vector as previously described [5]. The sequences were determined using an Automated Laser Fluorescence DNA Sequencer (ABI).
Phylogenetic analysis
The complete genomic sequence of the HN strain was submitted to GenBank (Access no. KF192951). The ORF sequences of the HN strain and 28 other flaviviruses from GenBank were included in phylogenetic analysis. Multiple alignments of polyprotein gene from GenBank were performed with the CLUSTAL W methods using the DNASTAR software package (DNASTAR version 6.0 Madison, WI, USA). Bootstrapped (1,000 times) neighbor-joining phylogenetic trees were generated by MEGA (version 3.1).
Cleavage site determination
Cleavage sites were identified using the proteolytic processing cascade scheme for flavivirus ORFs [6, 7]. Junctions between intracellular capsid and premembrane (Ci/prM), prM and envelope (prM/E), and E and nonstructural protein 1 (E/NS1), processed by the host cellular signalase, were determined on the basis of the highest cleavage potential score using the computer program Signal P-NN (http://www.cbs.dtu.dk/services/).
Secondary structures in 3′-UTR and cyclization sequence
The secondary structures of the 3′-UTR and cyclization sequence were investigated using the mfold program [8], based on the full-length 3′-terminal sequence following the NS5 gene stop codon [9].
Results
Features of the complete genome of the HN strain
The complete genome of the HN strain is 10,989 nucleotides in length. The 94-nucleotide 5′-UTR was of intermediate size and the 617-nucleotide 3′-UTR was quite long relative to those of other flaviviruses [10, 11]. The predicted long ORF (named ORF1 here) results in 3,425 residues (from 95 to 10,372 nt) having a molecular mass of about 380 kDa, which is similar to that of the BYD-1 (GenBank Access no. JF312912) and JS804 strains (JF895923). The ORF1 initiator AUG codon is flanked by an A at position −3 and a U at +4 [CAACUAUGU]. This should be considered a strong initiator since recent evidence has demonstrated that the base located at position +4 is not as important as first suggested, but that a purine (especially an adenine) at position −3 is characteristic of strong ribosome initiation sites [12, 13]. A second ORF (named ORF2 here) with an AUG located at nt 143, 48 bases downstream from the first AUG and, therefore, in-frame, with an A residue at −3 position and a C at +4 [UCAAUAUGC], should also be considered as having a strong initiator. Translation termination of the ORF1 and the ORF2 are represented by the same UAA.
The quantitative distribution of the four bases of ORF1 was found to be 28.63 % A, 28.92 % G, 22.48 % T, and 19.96 % C. The polyprotein is a slightly basic protein (PI 8.53) with a mainly hydrophobic N-terminus and a hydrophilic central region (data not shown). The secondary structure of the HN polyprotein was determined to possibly be as follows: 35 % of the residues form α-helices, 26 % form β-sheets, 20 % are within turns, and 20 % are in random coils.
Cleavage sites, potential glycosylation sites, and cysteine residues
The predicted cleavage sites of the HN strain are shown in Table 1. The N-termini of all sites expected to be cleaved by the viral serine protease (Cv/Ci, pr/M, NS2A/NS2B, NS3/NS4A, and NS4B/NS5) follow two C-terminal basic amino acids R(K)/R, as in most other mosquito-borne flaviviruses [14, 15]. Cleavage sites generating the N-termini of NS2B, NS3, NS4A, and NS5 are highly conserved among flaviviruses. The NS1/NS2A site, which is believed to be cleaved by an unknown cellular signalase, follows the sequence V-X-A (in which X is variable) [7]. The C-terminal quadrapeptide preceding the M/E cleavage site is PAYS in all mosquito-borne viruses. The predicted lengths of the potential structural and nonstructural proteins of the HN strain are as follows: C, 120 amino acids (aa); prM,177 aa; E, 501 aa; NS1, 342 aa; NS2A, 227 aa; NS2B, 131 aa; NS3, 619 aa; NS4A, 126 aa; 2K, 23 aa; NS4B, 254 aa; NS5, 905 aa.
The numbers of potential N-linked glycosylation sites in the prM, E, and NS1 genes in the HN strain follow a 2–2–3 pattern, which differs from that of the Bagaza virus. The patterns consisting of 12 cysteine residues in the E and NS1 genes and 6 cysteine residues clustered in the prM domain are found in all mosquito-borne flaviviruses.
3′-UTR secondary structure
Secondary structures of the 3′-UTR were predicted using the mfold program described previously [8]. Twenty-seven predicted folding patterns as well as folding energy levels were generated for the HN strain (Fig. 1). The long stable hairpin structure near the 3′-terminal sequence (3′-LSH) contains the conserved pentanucleotide (CACAG) in the loop. An octanucleotide (514CATATTGA521; called 3′-CYC) is involved in cyclization forms (Fig. 1). The consensus sequences generated from CS1 (512–532 nt), CS2 (389–411 nt), and CS3 (155–182 nt) of the HN strain are conserved across other flavivirus genomes [11, 15], while their organization (5′ to 3′) in the HN strain is CS3-CS2-CS1. Both CS2 and CS3 contribute to secondary structure.

The secondary structure of 3′-UTR of the HN strain as predicted by mfold program. Arrows indicate the 5′- and 3′-termini of the 3′-UTR. The 3′-UTR sequence is read downstream from the 5′-arrow clockwise along the partially double-strand structure
Phylogenetic relationships between the HN strain and other flaviviruses
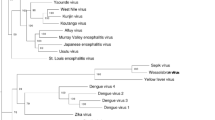
To evaluate evolutionary relationships between the HN strain virus and other flavivirus genus members, phylogenetic trees were constructed using polyprotein amino acid sequences. Some flaviviruses lack a complete genome sequence in GenBank; therefore, the 28 flavivirus complete genome sequences available in GenBank were used for phylogenetic analysis.
Phylogenetic analyses were performed using CLUSTAL W methods. The phylograms generated from polyprotein sequences (Fig. 2) reveal that all vector-borne viruses belong to two groups, a finding consistent with the well-established dichotomy between mosquito-borne and tick-borne viruses [16], and the HN strain grouped into the mosquito-borne virus clusters. Phylogenetic analysis of the polyprotein also showed that the HN strain is closely related to Tembusu virus and Bagaza virus (AY632545) (with 80.9 and 69.9 % identity, respectively), a zoonotic mosquito-borne flavivirus which causes both human and animal diseases [17, 18].

Phylogenetic relationships between the HN strain polyprotein and other flaviviruses
Discussion
In this study, we documented the complete genome sequence of the duck flavivirus HN strain. The length of ORF1 of the HN strain (3,425 aa) is one amino acid shorter than that of the Bagaza virus (3,426 aa). The HN 5′-UTR (94 nt) is the same length as that of the Bagaza virus, while the HN 3′-UTR (617 nt) is forty-eight bases longer than that of the Bagaza virus (566 nt) [14]. ORF1 and ORF2 start with different strong initiators and terminate at the same UAA stop codon, suggesting that the second AUG may also serve as a translation initiation site as described for dengue type 2 virus [1, 19]. Additional investigation should be conducted to identify whether proteins could be produced by this internal initiation.
The predicted cleavage sites of the HN strain basically follow the patterns established for other mosquito-borne viruses; likewise, cysteine residues are well conserved. The number of potential N-linked glycosylation sites is different from those reported for other flaviviruses, suggesting that N-linked glycosylation sites may be not related to epitope recognition [20]. The CS1 and CS2, shared by all mosquito-borne flaviviruses, are located 5′ to the putative 3′- terminal secondary structure [15]. Among the fully sequenced mosquito-borne flaviviruses, the HN strain is the second virus whose 3′-UTR has the CS3-CS2-CS1 organization (represented by Spondweni virus group).
Phylogenetic analysis shows that the HN strain is a novel flavivirus, most closely related to Bagaza virus of the Ntaya virus group. Thus far, there is no evidence that the duck flavivirus causes human disease. However, most flavivirus are zoonotic, suggesting they can be transmitted from animals to human beings. Evidence of bird infections by flaviviruses has been well documented, as in the case of West Nile virus, turkey meningoencephalitis virus, and Bagaza viruses, which cause both human and birds disease [17, 18, 21–25]. Even while the natural transmission mechanism of this Tembusu virus remains unknown, the potential for infection of human beings by this virus cannot be disregarded. Therefore, it will be necessary to monitor whether the novel duck Tembusu virus is transmitted from ducks to humans or vice versa. This can be achieved by developing duck Tembusu virus-specific serological and molecular diagnostics for testing human clinical specimens collected from the affected region. Much more epidemiological investigation is required to identify the transmission method of this virus. Eradication of flavivirus pathogens is unlikely because the viruses are maintained in animal reservoirs and can be transmitted by an unknown vector [26]. Therefore, vaccine development is necessary for prevention of this duck Tembusu virus.
In summary, this paper provides important information about the properties of this novel Tembusu virus, expands the available flavivirus sequences in public databases, and establishes the phylogenetic relationships between duck Tembusu virus and other flaviviruses. Features of the HN strain genome, polyprotein cleavage sites, 3′-UTR secondary structure, and 3′- sequence analysis have also been characterized.
References
B.D. Lindenbach, H.J. Thiel, C.M. Rice, in Fields virology, 5th edn., ed. by D.M. Knipe, P.M. Howley (Lippincott-Raven Publishers, Philadelphia, 2007), pp. 1101–1152
J.J. Su, S. Li, X.D. Hu, X.L. Yu, Y.Y. Wang, P.P. Liu, X.S. Lu, G.Z. Zhang, X.Y. Hu, D. Liu, X.X. Li, W.L. Su, H. Lu, N.S. Mok, P.Y. Wang, M. Wang, K.G. Tian, G.F. Gao, PLoS ONE 6(3), e18106 (2011)
Z.Z. Cao, C. Zhang, Y.H. Liu, W.C. Ye, J.W. Han, G.M. Ma, D.D. Zhang, F. Xu, X.H. Gao, Y. Tang, S.H. Shi, C. Zhang, B. He, M.J. Yang, X.H. Lu, Y. Huang, Y.X. Diao, X.J. Ma, D.D. Zhang, Emerg. Infect. Dis. 17(10), 1873–1875 (2011)
M. Liu, S.Y. Chen, Y.H. Chen, C.G. Liu, S.L. Chen, X.C. Yin, G. Li, Y. Zhang, J. Clin. Microbiol. 50(8), 2807–2809 (2012)
Y. Zhang, D.C. Guo, H.W. Geng, M. Liu, Q.L. Hu, J.W. Wang, G.Z. Tong, X.G. Kong, N.Y. Liu, C.G. Liu, Virus Res. 125, 42–53 (2007)
M. Liu, C.G. Liu, G. Li, X.J. Li, X.C. Yin, Y.H. Chen, Y. Zhang, J. Virol. 86(6), 3398–3399 (2012)
C.M. Rice, J.H. Strauss, Semin. Virol. 1, 357–367 (1990)
M. Zuker, D.H. Mathews, D.H Turner, In: Barciszewski J, Clark BFC (eds) Kluwer Dardsecht, 11–43(1999)
A.A. Khromykh, H. Meka, K.J. Guyatt, E.G. Westaway, J. Virol. 75, 6719–6728 (2001)
T.J. Chambers, C.S. Hahn, R. Galler, C.M. Rice, Annu. Rev. Microbiol. 44, 649–688 (1990)
V. Proutski, E.A. Gould, E.C. Holmes, Nucleic Acids Res. 25, 1194–1202 (1997)
M. Kozak, Nature 308, 241–246 (1984)
M. Kozak, J. Biol. Chem. 266, 19867–19870 (1991)
G. Kuno, J.J. Chang, Arch. Virol. 152, 687–696 (2007)
C.S. Hahn, Y.S. Hahn, C.M. Rice, E. Lee, L. Dalgarno, E.G. Strauss, J.H. Strauss, J. Mol. Biol. 198, 33–41 (1987)
G. Kuno, G.J. Chang, K.R. Tsuchiya, N. Karabatsos, C.B. Cropp, J. Virol. 72, 73–83 (1998)
E.A. Gould, T. Solomon, Lancet 371, 500–509 (2008)
M. Traore-Lamizana, H.G. Zeller, M. Mondo, J.P. Hervy, F. Adam, J. Med. Entomol. 31, 934–938 (1994)
G.R. Cleaves, J. Gen. Virol. 66, 2767–2771 (1985)
V. Vorndam, J.H. Mathews, A.D.T. Barrett, J.T. Roehrig, D.W. Trent, J. Gen. Virol. 74, 2653–2660 (1993)
R.J. Austin, T.L. Whiting, R.A. Anderson, M.A. Drebot, Can. Vet. J. 45, 117–123 (2004)
J.S. Mackenzie, D.J. Gubler, L.R. Petersen, Nat. Med. 10, 98–109 (2004)
D.E. Swayne, J.R. Beck, C.S. Smith, W.J. Shieh, S.R. Zaki, Emerg. Infect. Dis. 7, 751–753 (2001)
B.J. Barnard, J.H. Du Preez, S.P. Greyling, H.J. Venter, J. Vet. Res. 47, 89–94 (1950)
J.P. Digoutte, Am. J. Trop. Med. Hyg. 27, 376–377 (1978)
D.J. Gubler, Expert Rev. Vaccine 10(5), 563–565 (2011)
Acknowledgments
This study was supported by Grants from China Agriculture Research System (CARS-43-10), Chinese Special Fund for Agro-scientific Research in the Public Interest (201003012), and Natural Science Foundation of China (31072132).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Bai, X., Lv, R., Liu, C. et al. Molecular characterization of a duck Tembusu virus from China. Virus Genes 47, 478–482 (2013). https://doi.org/10.1007/s11262-013-0966-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-013-0966-3