
Abstract
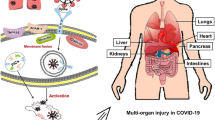
On December 30th 2019, some patients with pneumonia of unknown etiology were reported in the Program for Monitoring Emerging Diseases (ProMED), a program run by the International Society for Infectious Diseases (ISID), hypothesized to be related to subjects who had had contact with the seafood market in Wuhan, China. Chinese authorities instituted an emergency agency aimed at identifying the source of infection and potential biological pathogens. It was subsequently named by the World Committee on Virus Classification as 2019-nCoV (2019-novel coronavirus) or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A number of studies have demonstrated that 2019-nCoV and the SARS-CoV shared the same cell entry receptor named angiotensin-converting enzyme 2 (ACE2). This is expressed in human tissues, not only in the respiratory epithelia, but also in the small intestines, heart, liver, and kidneys. Here, we examine the most recent findings on the effects of SARS-CoV-2 infection on kidney diseases, mainly acute kidney injury, and the potential role of the chemokine network.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronaviruses are a family of pathogens infecting humans as well as other vertebrates and is, therefore, of great clinical and veterinary importance [1]. In 1983 Siddell et al. reported that Coronavirions are pleomorphic, generally spherical, 60 to 220 nm in diameter and are characterized by widely spaced, club-shaped surface projections about 20 nm in length [2]. Complete virions have a density in sucrose of approximately 1.18 g/ml [2]. The coronavirus genome is a linear molecule of single-stranded RNA. Additionally, the coronavirius nucleocapsid contains a non-glycosylated protein of 50,000 to 60,000 molecular weight [2]. It has been reported that Coronaviruses cause a large spectrum of diseases in humans and animals which can be largely categorized into two classes based on its respiratory or enteric tropisms [3]. It has been ascertained that their pathogenic potential ranges from respiratory and gastrointestinal diseases to hepatitis, encephalomyelitis, vasculitis and coagulopathies [4]. In several viral diseases, such as Dengue haemorrhagic fever, Yellow fever and some Human Immunodeficiency Virus (HIV) infection, an augmentation of pathology by the antibody response through deposition of immune complexes or antibody-mediated enhancement of infectivity. Chronic inflammation elicited by feline coronavirus infections represents an interesting virus-host system in which such mechanisms can be investigated. Several coronavirus-induced disease processes are driven or accompanied by immunopathological mechanisms (i.e. feline infectious peritonitis, a management problem in catteries). This antibody- and C’-mediated disease process displays interesting mechanistic parallels to other significant viral diseases in humans, such as hemorrhagic shock syndromes and damage to the lymphoreticular system. These viruses provide interesting experimental models by emulating complex disease processes and may represent an unrecognized threat in biomedical sciences. In contrast, murine coronavirus infections in rodents have been developed as helpful models for virus persistence and chronic infection [3, 5]. Late 2019, a report of a cluster with pneumonia of unknown etiology was reported and subsequently named by the World Committee on Virus Classification as 2019-nCoV or the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [6,7,8,9]. This novel coronavirus is hypothesized to be related to contact with a seafood market in Wuhan City, China [10]. In response, hospitals in the region held an emergency symposium, and federal agencies worked towards determining the source of infection and causative organism. On January 5th 2020, the World Health Organization (WHO) published a summary outlining their requests for further information from Chinese public health authorities, and reported a total of 44 patients with “pneumonia of unknown etiology” and 121 close contacts under surveillance. The WHO reported that 11 patients were severely ill, and many of affected those had had contact with the Huanan Seafood market [10]. Patients were reported to have symptoms such as fever, dyspnea and pulmonary infiltrates on chest radiographs. The virus predominantly spreads through respiratory droplets and close contact [9, 11,12,13] and concerningly, it has been reported that a large number of health care workers were infected during the initial phases of the pandemic in the city of Wuhan, China [11]. The virus is rapidly spreading from its origin to the rest of the world [14,15,16]. The rapidly expanding 2019-nCoV pandemic, caused by SARS-CoV, has challenged the medical community to an unprecedented degree [11]. Here, we briefly discuss the most recent findings on the main effects of SARS-CoV-2 infection on kidney diseases, mainly acute kidney injury, and the potential role of chemokine network.
SARS-CoV-2 and the kidney
It is known that 2019-nCoV predominantly affects the respiratory system with manifestations ranging from upper respiratory symptoms to acute respiratory distress syndrome (ARDS) [17]. Xu et al. [18] have investigated the clinical manifestations and imaging characteristics [computed tomography (CT)] in novel coronavirus pneumonia (NCP) caused by SARS-CoV-2, as disease defined on 8th February 2020 in China [18]. They concluded that CT imaging presentations of NCP are predominantly patchy ground-glass opacities in the peripheral zones under the pleura with partial consolidation, which will be reabsorbed with the formation of fibrotic strips. As a consequence, CT scan provides an important base for early diagnosis and treatment of NCP [19, 20]. It has been reported that patients with mild disease may have only low-grade fever and weakness without pneumonia, whereas severe patients will have dyspnea and/or hypoxemia, and those with critically severe illness will quickly progress to acute respiratory distress syndrome, septic shock, uncorrectable metabolic acidosis, coagulation dysfunction and death. Contrary to infection with SARS-CoV-2 and H7N9 avian influenza, which results in a high fever at the onset of infection, the initial symptoms of SARS-CoV-2 infection are atypical with only low-grade fever and long incubation period providing potent infective capabilities [21, 22].
Several studies have shown that 2019-nCoV and the SARS-CoV shared the same cell entry receptor, angiotensin-converting enzyme 2 (ACE2) [23]. It has been hypothesized that the ACE2 expression pattern in different organs, tissues, and cell types could be related to the risk pattern of 2019-nCoV infection [23]. It has been shown that ACE2 expression in humans is not only restricted to the epithelia of the lung, but also found in the small intestine, heart, liver, and kidneys [24]. Gu et al. [25] in autopsy specimens obtained from SARS affected patients found that immunohistochemical examination revealed SARS-CoV virions, RNA, and antigens in lung and other organs, including the kidney. Pacciarini et al. [26] established in vitro that association of SARS-CoV in proximal tubular epithelial cells in 31 cases showed persistent and productive infections, which were partially correlated with ACE2 expression. Moreover, using state-of-the-art single-cell techniques, Zou et al. [27] stratified organs into high and low risks according to the expression level of ACE2 concluding that the kidney should be listed as high risk. These findings indicate the possibility of 2019-nCoV infections in kidney cells. Epidemiological data indicate that over two-thirds of patients who died from 2019-nCoV had diabetes or cardiovascular disease [28]. Most patients with these diseases undergo to angiotensin-receptor blockers (ARBs) treatment. Several studies have shown that ARBs are able to increase ACE2 expression in the kidney and the heart [29]. It has been shown that SARS-CoV-2 entry into target cells is a highly regulated multi-step processes, of which binding to ACE2 is merely the first. TMPRSS2 (Transmembrane Serine Protease 2) is an essential serine protease required for spike glycoprotein priming after binding to ACE2. Increased expression of ACE2 by ARBs may induce the sequestration of SARS-CoV-2 in the cell membrane; however, this may not be paralleled by an increase in TMPRSS2, which could act in limiting viral infection. Additionally, membrane-bound ACE2 is processed by the metalloproteinase ADAM17, which cleaves the ACE2 ectodomain to allow the release of a soluble form. Although the overall effects on urological patients of delayed treatment and diagnosis remain to be defined, Perico et al. have reported that the available epidemiological data indicate that acute kidney injury (AKI) is one of the main indicators of prognosis of 2019-nCoV, and diabetes is the main renal co-morbidity [29]. The potential impact of 2019-nCoV on patients affected by other renal conditions, such as end-stage renal disease and transplantation, is still not clear at this phase of the pandemic [29]. Interestingly, analysis of peripheral blood samples from hemodialysis patients infected with SARS-CoV-2 showed a remarkable reduction in the numbers of T-lymphocytes, T-helper cells, and Natural Killer (NK) T-lymphocytes, as well as lower serum levels of inflammatory cytokines, compared to non-hemodialysis patients with 2019-nCoV. Collectively, this study reported that hemodialysis patients with 2019-nCoV are more likely to experience mild disease and lower rates of progression towards pneumonia, likely secondary, to the reduced immune function and decreased cytokine storms [29]. Additionally, the risk of acquiring 2019-nCoV from organ donation is low. However, it has been shown that SARS-CoV-2 has a high tropism for the kidney, where it has been shown to replicate in almost 30% of 2019-nCoV 19 patients. For this reason, screening for 2019-nCoV in kidney donors should be strongly considered. Additionally, living donors who have clinical manifestations or have travelled to high-risk areas are advised to postpone donation for 14–28 days. Separately, Nacker et al. concluded that in previous reports SARS and Middle East respiratory syndrome-corona virus (MERS-CoV) infections, indicate that AKI developed in 5% to 15% cases and carried a high (60–90%) mortality rate. Early reports suggest a lower incidence (3–9%) of AKI in those with 2019-nCoV infection [30]. The pathophysiological bases related to kidney involvement are still unclear. Hypothesized mechanisms include sepsis leading to cytokine storm syndrome or direct cellular injury due to the virus. Angiotensin-converting enzyme and dipeptidyl peptidase-4, both expressed on renal tubular cells, were identified as binding partners for SARS-CoV and MERS-CoV, respectively. Viral RNA has been identified in kidney tissue and urine in both infections. Recently SARS-CoV-2 has also been isolated from the urine samples of an infected patient, suggesting the kidney as target of this novel coronavirus [30].
Chemokine network and 2019-nCoV
Inflammation in viral pneumonia can be a double-edged sword. Although inflammation is important for local tissues to combat infection, increased inflammatory responses in pneumonia patients result in the excessive release of pro-inflammatory cytokines known as a “cytokine storm”, leading to detrimental outcomes, such as diffuse alveolar damage, fibrosis, progressive respiratory failure and multiple organ dysfunction [31]. 2019-nCoV patients show a dramatic cytokine storm, which is in reminiscent of previous SARS-CoV infections [32]. A higher number of clinical studies have suggested that a cytokine storm is associated with 2019-nCoV severity and is also a crucial cause of death from 2019-nCoV [33]. It has been reported that nearly 14% of infected patients develop severe disease requiring hospitalization and oxygen support and that 5% require admission to an intensive care unit [34]. 2019-nCoV mild patients demonstrate a high level of IL1-β, IFN-γ, CXCL10/IP-10 and CCL2/MCP-1, whilst patients requiring Intensive Care Unit admission have higher levels of G-CSF, CXCL10/IP-10, CCL2/MCP-1 and CCL3/MIP-1ɑ. A specific inflammatory cytokine and chemokine expression profile have been identified in 2019nCoV patients. Additionally, autopsy lung tissue has shown diffuse infiltration of hyperactivated T-lymphocytes [34, 35]. In a study by Xiong et al. [36] an expression of a large number of cytokines was found to be significantly elevated in 2019-nCoV patients’ bronchoalveolar lavage fluid samples compared to control, including pro-inflammatory cytokines CXCL1, CXCL2, CXCL6, CXCL8, CXCL10/IP-10, CCL2/MCP-1, CCL3/MIP-1A and CCL4/MIP1B. These data suggest that SARS-CoV-2 virus infection led to cytokine storm, which correlated with disease severity. Recently, La Rosée et al. reported that using a newly developed 2019-nCoV Inflammation Score (CIS), patients were prospectively stratified for targeted inhibition of cytokine signaling by ruxolitinib, a drug approved as a potent and selective inhibitor of Janus kinase 1 and 2 for the treatment of primary myelofibrosis, post polycythemia vera or post-essential trombocytemia myelofibrosis, all highly inflammatory conditions. Patients were treated with efficacy/toxicity guided step-up dosing up to 14 days. Out of 105 patients treated between March 30th and April 15th, 2020, 14 patients with a CIS ≥ 10 out of 16 points received ruxolitinib over a median of 9 days with a median cumulative dose of 135 mg. A total of 12 out of 14 patients achieved significant reduction of CIS by ≥ 25% on day 7 with sustained clinical improvement in 11 out of 14 patients without short term red flag warnings of Ruxinduced toxicity [34]. Ruxolitinib treatment in affected patients with hyperinflammation has been found to be safe with signals of efficacy for cytokine release syndrome-intervention to prevent or overcome multiorgan failure. Concomitantly, the same Authors have initiated a multicenter phase-II clinical trial (NCT04338958). Importantly, the study by La Rosée et al. [34] was aimed at diminishing or stopping the detrimental cytokine signaling, therefore, preventing further organ damages. Increased transcription of the respective chemokines receptors such as CCR2 (CCL2/MCP-1 receptor) and CCR5 (CCL3/MIP-1ɑ receptor) was also observed, indicating the activation of these inflammatory signals. Chemokines and their conventional and atypical receptors play an important role in the leukocyte migration and activation of immune cells at the sites of infection [37, 38]. It has been shown that CCR2 and CCR5 deficient mice infected with mouse-adapted SARS-CoV virus exhibited defects in directing inflammatory cells to the airway, causing severe disease and increased mortality [39]. In addition, high levels of macrophages chemoattractants CXCL10/IP-10 and CCL2/MCP-1 and neutrophil chemoattractants CXCL2 and CXCL8 facilitate the migration of these immune cells to the site of infection, which was consistent with mononuclear cell infiltrates in lung tissues of 2019-nCoV patients [35].
Chemokines network, 2019-nCoV and kidney diseases
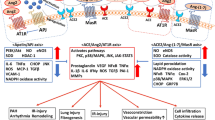
AKI defines a systemic disease characterized by acute loss of renal function and the storage of end products of nitrogen metabolism. Ischemic AKI is the most common cause of AKI, and inflammatory responses are inevitably involved in ischemic AKI. In the process of ischemic AKI, various factors (among them the so-called chemokines) are responsible for promoting and recruiting immune cells to the injured kidney [40].
The RUBY study was a multi-center international prospective observational study, aimed at identifying the biomarkers of persistent stage 3 AKI as defined by the KDIGO criteria [41]. In this large international cohort of surgical and medical intensive care patients, it has been shown that several candidate molecules predicted the persistence of AKI. However the most promising was a previously unknown candidate called urinary chemokine C–C motif ligand 14 (CCL14). Among patients who did not persist at any stage of AKI, the urinary CCL14 concentrations within different comorbid conditions were similar suggest that urinary CCL14 elevations were specific to AKI persistence. Given this performance, CCL14 could be an important mediator of renal tissue damage and non-recovery, providing potential novel diagnostic and prognostic information that is important for the management of patients with AKI, including decisions related to initiation of renal replacement therapy. Additionally, CCL14 has been shown to be an inflammatory marker, which identifies the risk of developing the end-stage renal disease in diabetics [42]. CCL14 is a member of the chemokine family that has been recognized its crucial roles in monocyte/macrophage recruitment and the tissue injury and repair processes. It is associated in a variety of diseases including rheumatoid arthritis, multiple sclerosis, and lupus [43, 44]. CCL14 binds to the conventional CC chemokine receptors 1, 5 and 3 (CCR1, CCR5 and CCR3) [45], as well as to the atypical chemokine receptor ACKR2 [46]. Today, little is known regarding the role of CCL14 in AKI, mainly because CCL14 it cannot be studied in pre-clinical models. As macrophage recruitment and polarization are accepted as important players in kidney tissue damage and the development of persistent kidney dysfunction [47], a putative role for CCL14 can be hypothesized. Briefly, in a pro-inflammatory environment, TNF-α and other inflammatory mediators are released from injured epithelium and bind to TNF receptors, leading to release of CCL14 from tubular epithelial cells. The binding of CCL14 to CCR1 and CCR5 receptors on monocytes and T-lymphocytes induces chemotaxis towards the site of injury, where monocytes differentiate into macrophages and naïve T-lymphocytes differentiate into pro-inflammatory T-lymphocytes helper 1 (Th1) that are pathogenic and can extend and magnify tissue damage.
C–C motif ligand 1 (CCL2 also known as Monocyte Chemotactic Protein-1 MCP1) is a member of the chemokine family. CCL2 binds to conventional CC chemokine receptor 2 (CCR2) on monocytes and regulates the trafficking of monocytes from bone marrow to tissues in response to inflammatory signals. Interestingly, similar to CCL14, fine tuning of the bioavailability of CCL2 relies on its binding to the atypical chemokine receptor ACKR2 [46], confirming the crucial role of this chemokine receptor and its pro-inflammatory ligands in AKI. It has been shown that mRNA expression is increased in ischemia–reperfusion injury, and thus, it has value as a biomarker for mononuclear inflammatory processes that occur after ischemic AKI [48]. In further studies, CCL2 was found to be a potent chemokine produced by kidney cells, acting as a mediator in acute ischemic and toxic kidney injuries. The potential utility of CCL2 as a biomarker was also supported by clinical assessments observing increases in urinary CCL2 protein in patients with AKI. It has been shown that CCL2 increases to a greater extent than Neutrophil Gelatinase-Associated Lipocalin (NGAL) in intrarenal injury, although NGAL and CCL2 gene expression increase comparably in renal injury. The NGAL gene is induced by uremia per se in the absence of renal injury, while the CCL2 gene not. Thus, it could provide complementary information to that provided by NGAL [49]. Finally, elevated plasma CCL2 has been associated with an increased risk of AKI and death after cardiac operations [50].
In a model of dextran sodium sulfate colitis, chemokine C-X-C motif ligand 1 (CXCL1) has been shown to be the major mediator of colon-kidney organ crosstalk [51]. Chemokine C-X-C motif ligand 8 (CXCL8, also known as IL-8), the human homolog of rodent CXCL1, is an early pro-inflammatory chemokine secreted by mononuclear immune cells and epithelial cells to trigger neutrophil chemotaxis and activation by binding and activating conventional CXC chemokine receptors 1 and 2 (CXCR1 and CXCR2) [52]. CXCL8/CXCL1 has been implicated in a broad range of diseases characterized by polymorphonuclear neutrophils cell infiltration, including AKI [53]. If the initial role of the CXCL8 pathway is involved in the local innate immune response after activation of the NF-κB pathway, persistent or up-regulated expression of CXCL8 could preserve immune cell recruitment, with deleterious effects on inflamed tissue. In a recent study of Amrouche et al. it has been found that miR-146a targets the NF-κB pathway in tubular cells and consequently, regulate CXCL8 secretion. Deletion of miR-146a in vivo enhanced the formation of tubular lesions after unilateral AKI, the infiltration of immune cells, and the development of renal fibrosis [54].
The expression of C-X3-C motif ligand 1 (CX3CL1, also known as fractalkine) induced by Lipopolysaccharides (LPS) is also one of the pivotal inflammatory elements linked to renal tissue damage. CX3CL1 participates in the inflammatory response in several biological systems by binding to its receptor CX3CR1. Most studies demonstrated its role in promoting renal pathopoiesis (glomerular inflammation and endothelial injury); however, several recent studies showed that it could also reduce renal pathopoiesis. Thus, to date the CX3CL1/CX3CR1 axis is considered to be a double-edged sword that could provide novel perspectives into the pathogenesis and treatment of renal diseases and disorders [55]. Furthermore, Park et al. found that LPS can up-regulate the expression of CXC3CL1 and can contribute to renal inflammation leading to chronic renal allograft rejection. A recent study revealed that LPS can cause inflammatory responses both in vitro and in vivo of glomerular podocytes by means of enhancing CX3CL1 and activating the Wnt/β-catenin signaling pathway in podocytes, resulting in a decrease in the expression of podocyte-specific mRNA and proteins and is involved in the occurrence and development of AKI [56].
The bioavailability of chemokines represents a crucial hub in AKI. The crucial role exerted by pro-inflammatory chemokines such as CCL14 and CCL2 suggest a key contribution of ACKR2 in AKI pathogenesis. The atypical chemokine receptor ACKR2 represents a scavenger receptor, which binds and sequesters many inflammatory CC chemokines but does not transduce typical G-protein mediated signaling events [57]. Human biopsies stained with antibodies raised against-ACKR2 revealed increased staining in the diabetic kidney, especially in some tubules, interstitial cells, leukocytes, and endothelial cells [58]. Accordingly, the established ACKR2 knockout mouse exhibits several features of human diabetic nephropathy and extensive renal inflammation [58]. A recent report also showed that ACKR2 limits leukocyte infiltration (mainly monocytes, T-lymphocytes, and Ly6Chi inflammatory macrophages), inflammation, and fibrotic tissue remodeling after AKI, thus preventing progression to chronic kidney disease [59]. Furthermore, it has been demonstrated that ACKR2 plays an important role in limiting glomerular and tubulointerstitial injury, inflammation, and fibrotic remodeling due to scavenging of chemokines in the tubulointerstitial compartment [60]. Collectively, these data identify ACKR2 as a potential target for therapeutic approaches in renal inflammatory and fibrotic disease associated with AKI.
Conclusions
It has been ascertained that antiviral drugs administered shortly after symptom onset can reduce infectiousness by reducing viral shedding in the respiratory secretions of patients, and targeted prophylactic treatment of contacts could reduce their risk of becoming infected [61]. Yang et al. entitled their manuscript “Diagnosis and treatment of 2019-nCoV: acute kidney injury cannot be ignored”, a peer-reviewed article [62]. As suggested by Zhang and Liang [63] particular attention to renal function should be taken into account when treating 2019-nCoV patients [64]. Such information calls for patient care regarding the renal function of patients currently under emergency and potential post-cure treatment for kidney recovery. Furthermore, several authors have published regarding patients affected by diabetes [65,66,67,68,69,70,71,72], and those undergoing kidney transplant [73,74,75,76]. In particular, Muniyappa et al. [71] discussed potential mechanisms by which diabetes modulates host-viral interactions and host-immune responses. They found that although studies exist in humanized ACE2 mice and non-human primates aimed at understanding how hyperglycemia, hyperinsulinemia, and hypoglycemic agents affect the pathogenesis of 2019-nCoV, studies regarding how diabetes mellitus affects the efficacy of vaccines and anti-viral investigational agents currently in trials are warranted. Furthermore, Naspro and Del Pozzo stated: “In particular, urologists manage many patients with oncological diseases and surgical priorities, and also many non-oncological life-threatening conditions and other disorders that only affect the quality of life.” [77]. Furthermore, the actual knowledge suggests a particular attention to subjects with a unique kidney as more prone to kidney failure [78].
References
Lai MM (1990) Coronavirus: organization, replication and expression of genome. Annu Rev Microbiol 44:303–333. https://doi.org/10.1146/annurev.mi.44.100190.001511
Siddell S, Wege H, Ter Meulen V (1983) The biology of coronaviruses. J General Virol 64(Pt 4):761–776. https://doi.org/10.1099/0022-1317-64-4-761
Compton SR, Barthold SW, Smith AL (1993) The cellular and molecular pathogenesis of coronaviruses. Lab Anim Sci 43(1):15–28
Wege H (1995) Immunopathological aspects of coronavirus infections. Springer Semin Immunopathol 17(2–3):133–148. https://doi.org/10.1007/bf00196162
Wege H, Siddell S, ter Meulen V (1982) The biology and pathogenesis of coronaviruses. Curr Top Microbiol Immunol 99:165–200. https://doi.org/10.1007/978-3-642-68528-6_5
Conway MJ (2020) Identification of coronavirus sequences in carp cDNA from Wuhan, China. J Med Virol. https://doi.org/10.1002/jmv.25751
Coronaviridae Study Group of the International Committee on Taxonomy of V (2020) The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5(4):536–544. https://doi.org/10.1038/s41564-020-0695-z
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y, Li Y, Wang X, Peng Z (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan. JAMA, China. https://doi.org/10.1001/jama.2020.1585
Zhou M, Zhang X, Qu J (2020) Coronavirus disease 2019 (COVID-19): a clinical update. Front Med. https://doi.org/10.1007/s11684-020-0767-8
Bogoch II, Watts A, Thomas-Bachli A, Huber C, Kraemer MUG, Khan K (2020) Pneumonia of unknown aetiology in Wuhan, China: potential for international spread via commercial air travel. J Travel Med. https://doi.org/10.1093/jtm/taaa008
Givi B, Schiff BA, Chinn SB, Clayburgh D, Iyer NG, Jalisi S, Moore MG, Nathan CA, Orloff LA, O’Neill JP, Parker N, Zender C, Morris LGT, Davies L (2020) Safety recommendations for evaluation and surgery of the head and neck during the COVID-19 pandemic. JAMA. https://doi.org/10.1001/jamaoto.2020.0780
Legido-Quigley H, Mateos-Garcia JT, Campos VR, Gea-Sanchez M, Muntaner C, McKee M (2020) The resilience of the Spanish health system against the COVID-19 pandemic. Lancet Publ Health. https://doi.org/10.1016/S2468-2667(20)30060-8
Lescure FX, Bouadma L, Nguyen D, Parisey M, Wicky PH, Behillil S, Gaymard A, Bouscambert-Duchamp M, Donati F, Le Hingrat Q, Enouf V, Houhou-Fidouh N, Valette M, Mailles A, Lucet JC, Mentre F, Duval X, Descamps D, Malvy D, Timsit JF, Lina B, van-der-Werf S, Yazdanpanah Y (2020) Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect Dis. https://doi.org/10.1016/S1473-3099(20)30200-0
Garcia-Castrillo L, Petrino R, Leach R, Dodt C, Behringer W, Khoury A, Sabbe M (2020) European Society For Emergency Medicine position paper on emergency medical systems response to COVID-19. Eur J Emerg Med. https://doi.org/10.1097/MEJ.0000000000000701
Singhal T (2020) A review of coronavirus disease-2019 (COVID-19). Indian J Pediatr 87(4):281–286. https://doi.org/10.1007/s12098-020-03263-6
Wang C, Horby PW, Hayden FG, Gao GF (2020) A novel coronavirus outbreak of global health concern. Lancet 395(10223):470–473. https://doi.org/10.1016/S0140-6736(20)30185-9
Sundaram M, Ravikumar N, Bansal A, Nallasamy K, Basavaraja GV, Lodha R, Gupta D, Odena MP, Ashwath RNR, Jayashree M, For Intensive Care Chapter Of Indian Academy Of P (2020) Novel coronavirus 2019 (2019-nCoV) infection: part II - respiratory support in the pediatric intensive care unit in resource-limited settings. Indian Pediatr 57(4):335–342
Xu YH, Dong JH, An WM, Lv XY, Yin XP, Zhang JZ, Dong L, Ma X, Zhang HJ, Gao BL (2020) Clinical and computed tomographic imaging features of novel coronavirus pneumonia caused by SARS-CoV-2. J Infect 80(4):394–400. https://doi.org/10.1016/j.jinf.2020.02.017
Agostini A, Floridi C, Borgheresi A, Badaloni M, Esposto Pirani P, Terilli F, Ottaviani L, Giovagnoni A (2020) Proposal of a low-dose, long-pitch, dual-source chest CT protocol on third-generation dual-source CT using a tin filter for spectral shaping at 100 kVp for CoronaVirus Disease 2019 (COVID-19) patients: a feasibility study. Radiol Med (Torino). https://doi.org/10.1007/s11547-020-01179-x
Li B, Li X, Wang Y, Han Y, Wang Y, Wang C, Zhang G, Jin J, Jia H, Fan F, Ma W, Liu H, Zhou Y (2020) Diagnostic value and key features of computed tomography in coronavirus disease 2019. Emerg Microbes Infect. https://doi.org/10.1080/22221751.2020.1750307
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, Hui DSC, Du B, Li LJ, Zeng G, Yuen KY, Chen RC, Tang CL, Wang T, Chen PY, Xiang J, Li SY, Wang JL, Liang ZJ, Peng YX, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu JY, Chen Z, Li G, Zheng ZJ, Qiu SQ, Luo J, Ye CJ, Zhu SY, Zhong NS, China Medical Treatment Expert Group for C (2020) Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. https://doi.org/10.1056/NEJMoa2002032
Liu W, Zhang Q, Chen J, Xiang R, Song H, Shu S, Chen L, Liang L, Zhou J, You L, Wu P, Zhang B, Lu Y, Xia L, Huang L, Yang Y, Liu F, Semple MG, Cowling BJ, Lan K, Sun Z, Yu H, Liu Y (2020) Detection of Covid-19 in children in early January 2020 in Wuhan, China. N Engl J Med 382(14):1370–1371. https://doi.org/10.1056/NEJMc2003717
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273. https://doi.org/10.1038/s41586-020-2012-7
Li W, Wong SK, Li F, Kuhn JH, Huang IC, Choe H, Farzan M (2006) Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J Virol 80(9):4211–4219. https://doi.org/10.1128/JVI.80.9.4211-4219.2006
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202(3):415–424. https://doi.org/10.1084/jem.20050828
Pacciarini F, Ghezzi S, Canducci F, Sims A, Sampaolo M, Ferioli E, Clementi M, Poli G, Conaldi PG, Baric R, Vicenzi E (2008) Persistent replication of severe acute respiratory syndrome coronavirus in human tubular kidney cells selects for adaptive mutations in the membrane protein. J Virol 82(11):5137–5144. https://doi.org/10.1128/JVI.00096-08
Zou X, Chen K, Zou J, Han P, Hao J, Han Z (2020) Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. https://doi.org/10.1007/s11684-020-0754-0
Jessup JA, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell MC, Ferrario CM (2006) Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circul Physiol 291(5):H2166–2172. https://doi.org/10.1152/ajpheart.00061.2006
Perico L, Benigni A, Remuzzi G (2020) Should COVID-19 Concern nephrologists? Why and to what extent? The emerging impasse of angiotensin blockade. Nephron. https://doi.org/10.1159/000507305
Naicker S, Yang CW, Hwang SJ, Liu BC, Chen JH, Jha V (2020) The novel coronavirus 2019 epidemic and kidneys. Kidney Int. https://doi.org/10.1016/j.kint.2020.03.001
Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB (2014) Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Natl Acad Sci USA 111(10):3799–3804. https://doi.org/10.1073/pnas.1400593111
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Hu B, Huang S, Yin L (2020) The cytokine storm and COVID-19. J Med Virol. https://doi.org/10.1002/jmv.26232
La Rosee F, Bremer HC, Gehrke I, Kehr A, Hochhaus A, Birndt S, Fellhauer M, Henkes M, Kumle B, Russo SG, La Rosee P (2020) The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia. https://doi.org/10.1038/s41375-020-0891-0
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L, Tai Y, Bai C, Gao T, Song J, Xia P, Dong J, Zhao J, Wang FS (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8(4):420–422. https://doi.org/10.1016/S2213-2600(20)30076-X
Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, Guo D, Hu W, Yang J, Tang Z, Wu H, Lin Y, Zhang M, Zhang Q, Shi M, Liu Y, Zhou Y, Lan K, Chen Y (2020) Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 9(1):761–770. https://doi.org/10.1080/22221751.2020.1747363
Borroni EM, Savino B, Bonecchi R, Locati M (2018) Chemokines sound the alarmin: the role of atypical chemokine in inflammation and cancer. Semin Immunol 38:63–71. https://doi.org/10.1016/j.smim.2018.10.005
Griffith JW, Sokol CL, Luster AD (2014) Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 32:659–702. https://doi.org/10.1146/annurev-immunol-032713-120145
Sheahan T, Morrison TE, Funkhouser W, Uematsu S, Akira S, Baric RS, Heise MT (2008) MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog 4(12):e1000240. https://doi.org/10.1371/journal.ppat.1000240
Zheng L, Gao W, Hu C, Yang C, Rong R (2019) Immune cells in ischemic acute kidney injury. Curr Protein Pept Sci 20(8):770–776. https://doi.org/10.2174/1389203720666190507102529
Hoste E, Bihorac A, Al-Khafaji A, Ortega LM, Ostermann M, Haase M, Zacharowski K, Wunderink R, Heung M, Lissauer M, Self WH, Koyner JL, Honore PM, Prowle JR, Joannidis M, Forni LG, Kampf JP, McPherson P, Kellum JA, Chawla LS, Investigators R (2020) Identification and validation of biomarkers of persistent acute kidney injury: the RUBY study. Intens Care Med. https://doi.org/10.1007/s00134-019-05919-0
Niewczas MA, Pavkov ME, Skupien J, Smiles A, Md Dom ZI, Wilson JM, Park J, Nair V, Schlafly A, Saulnier PJ, Satake E, Simeone CA, Shah H, Qiu C, Looker HC, Fiorina P, Ware CF, Sun JK, Doria A, Kretzler M, Susztak K, Duffin KL, Nelson RG, Krolewski AS (2019) A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med 25(5):805–813. https://doi.org/10.1038/s41591-019-0415-5
Rump L, Mattey DL, Kehoe O, Middleton J (2017) An initial investigation into endothelial CC chemokine expression in the human rheumatoid synovium. Cytokine 97:133–140. https://doi.org/10.1016/j.cyto.2017.05.023
Vyshkina T, Sylvester A, Sadiq S, Bonilla E, Perl A, Kalman B (2008) CCL genes in multiple sclerosis and systemic lupus erythematosus. J Neuroimmunol 200(1–2):145–152. https://doi.org/10.1016/j.jneuroim.2008.05.016
Detheux M, Standker L, Vakili J, Munch J, Forssmann U, Adermann K, Pohlmann S, Vassart G, Kirchhoff F, Parmentier M, Forssmann WG (2000) Natural proteolytic processing of hemofiltrate CC chemokine 1 generates a potent CC chemokine receptor (CCR)1 and CCR5 agonist with anti-HIV properties. J Exp Med 192(10):1501–1508. https://doi.org/10.1084/jem.192.10.1501
Vacchini A, Locati M, Borroni EM (2016) Overview and potential unifying themes of the atypical chemokine receptor family. J Leukoc Biol 99(6):883–892. https://doi.org/10.1189/jlb.2MR1015-477R
Meng XM, Tang PM, Li J, Lan HY (2015) Macrophage phenotype in kidney injury and repair. Kidney Dis 1(2):138–146. https://doi.org/10.1159/000431214
Beker BM, Corleto MG, Fieiras C, Musso CG (2018) Novel acute kidney injury biomarkers: their characteristics, utility and concerns. Int Urol Nephrol 50(4):705–713. https://doi.org/10.1007/s11255-017-1781-x
Urbschat A, Obermuller N, Paulus P, Reissig M, Hadji P, Hofmann R, Geiger H, Gauer S (2014) Upper and lower urinary tract infections can be detected early but not be discriminated by urinary NGAL in adults. Int Urol Nephrol 46(12):2243–2249. https://doi.org/10.1007/s11255-014-0831-x
Moledina DG, Isguven S, McArthur E, Thiessen-Philbrook H, Garg AX, Shlipak M, Whitlock R, Kavsak PA, Coca SG, Parikh CR, Translational Research Investigating Biomarker Endpoints in Acute Kidney Injury C (2017) Plasma monocyte chemotactic protein-1 is associated with acute kidney injury and death after cardiac operations. Ann Thoracic Surg 104(2):613–620. https://doi.org/10.1016/j.athoracsur.2016.11.036
Ranganathan P, Jayakumar C, Manicassamy S, Ramesh G (2013) CXCR2 knockout mice are protected against DSS-colitis-induced acute kidney injury and inflammation. Am J Physiol Renal Physiol 305(10):F1422–1427. https://doi.org/10.1152/ajprenal.00319.2013
Kobayashi Y (2008) The role of chemokines in neutrophil biology. Front Biosci 13:2400–2407. https://doi.org/10.2741/2853
Cugini D, Azzollini N, Gagliardini E, Cassis P, Bertini R, Colotta F, Noris M, Remuzzi G, Benigni A (2005) Inhibition of the chemokine receptor CXCR2 prevents kidney graft function deterioration due to ischemia/reperfusion. Kidney Int 67(5):1753–1761. https://doi.org/10.1111/j.1523-1755.2005.00272.x
Amrouche L, Desbuissons G, Rabant M, Sauvaget V, Nguyen C, Benon A, Barre P, Rabate C, Lebreton X, Gallazzini M, Legendre C, Terzi F, Anglicheau D (2017) MicroRNA-146a in human and experimental ischemic AKI: CXCL8-dependent mechanism of action. J Am Soc Nephrol 28(2):479–493. https://doi.org/10.1681/ASN.2016010045
Zhuang Q, Cheng K, Ming Y (2017) CX3CL1/CX3CR1 axis, as the therapeutic potential in renal diseases: friend or foe? Curr Gene Ther 17(6):442–452. https://doi.org/10.2174/1566523218666180214092536
Senouthai S, Wang J, Fu D, You Y (2019) Fractalkine is involved in lipopolysaccharide-induced podocyte injury through the wnt/beta-catenin pathway in an acute kidney injury mouse model. Inflammation 42(4):1287–1300. https://doi.org/10.1007/s10753-019-00988-1
Cancellieri C, Vacchini A, Locati M, Bonecchi R, Borroni EM (2013) Atypical chemokine receptors: from silence to sound. Biochem Soc Trans 41(1):231–236. https://doi.org/10.1042/BST20120246
Zheng S, Coventry S, Cai L, Powell DW, Jala VR, Haribabu B, Epstein PN (2016) Renal protection by genetic deletion of the atypical chemokine receptor ACKR2 in diabetic OVE mice. J Diabetes Res 2016:5362506. https://doi.org/10.1155/2016/5362506
Lux M, Blaut A, Eltrich N, Bideak A, Muller MB, Hoppe JM, Grone HJ, Locati M, Vielhauer V (2019) The atypical chemokine receptor 2 limits progressive fibrosis after acute ischemic kidney injury. Am J Pathol 189(2):231–247. https://doi.org/10.1016/j.ajpath.2018.09.016
Bideak A, Blaut A, Hoppe JM, Muller MB, Federico G, Eltrich N, Grone HJ, Locati M, Vielhauer V (2018) The atypical chemokine receptor 2 limits renal inflammation and fibrosis in murine progressive immune complex glomerulonephritis. Kidney Int 93(4):826–841. https://doi.org/10.1016/j.kint.2017.11.013
Mitja O, Clotet B (2020) Use of antiviral drugs to reduce COVID-19 transmission. Lancet Global Health. https://doi.org/10.1016/S2214-109X(20)30114-5
Yang XH, Sun RH, Chen DC (2020) Diagnosis and treatment of COVID-19: acute kidney injury cannot be ignored. Zhonghua yi xue za zhi 100:E017. https://doi.org/10.3760/cma.j.cn112137-20200229-00520
Zhang F, Liang Y (2020) The potential risk of kidney vulnerable to novel coronavirus 2019 infection. Am J Physiol Renal Physiol. https://doi.org/10.1152/ajprenal.00085.2020
Gabrielson AT, Kohn TP, Clifton MM (2020) COVID-19 and the urology match: perspectives and a call to action. J Urol. https://doi.org/10.1097/ju.0000000000001037
Extance Andy (2020) Covid-19 and long term conditions: what if you have cancer, diabetes, or chronic kidney disease? BMJ 368:1270. https://doi.org/10.1136/bmj.m1270
Basile C, Combe C, Pizzarelli F, Covic A, Davenport A, Kanbay M, Kirmizis D, Schneditz D, van der Sande F, Mitra S (2020) Recommendations for the prevention, mitigation and containment of the emerging SARS-CoV-2 (COVID-19) pandemic in haemodialysis centres. Nephrol Dial Transpl. https://doi.org/10.1093/ndt/gfaa069
Burgner A, Ikizler TA, Dwyer JP (2020) COVID-19 and the Inpatient Dialysis Unit: Managing Resources During Contingency Planning Pre-Crisis. Clin J Am Soc Nephrol, CJASN. https://doi.org/10.2215/CJN.03750320
Extance A (2020) Covid-19 and long term conditions: what if you have cancer, diabetes, or chronic kidney disease? BMJ 368:m1174. https://doi.org/10.1136/bmj.m1174
Ferrey AJ, Choi G, Hanna RM, Chang Y, Tantisattamo E, Ivaturi K, Park E, Nguyen L, Wang B, Tonthat S, Rhee CM, Reddy U, Lau WL, Huang SS, Gohil S, Amin AN, Hsieh L, Cheng TT, Lee RA, Kalantar-Zadeh K (2020) A case of novel coronavirus disease 19 in a chronic hemodialysis patient presenting with gastroenteritis and developing severe pulmonary disease. Am J Nephrol. https://doi.org/10.1159/000507417
Kliger AS, Silberzweig J (2020) Mitigating risk of COVID-19 in dialysis facilities. Clin J Am Soc Nephrol, CJASN. https://doi.org/10.2215/CJN.03340320
Muniyappa R, Gubbi S (2020) COVID-19 pandemic, corona viruses, and diabetes mellitus. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00124.2020
Wang R, Liao C, He H, Hu C, Wei Z, Hong Z, Zhang C, Liao M, Shui H (2020) COVID-19 in hemodialysis patients: a report of 5 cases. Am J Kidney Dis. https://doi.org/10.1053/j.ajkd.2020.03.009
Gandolfini I, Delsante M, Fiaccadori E, Zaza G, Manenti L, Degli Antoni A, Peruzzi L, Riella LV, Cravedi P, Maggiore U (2020) COVID-19 in kidney transplant recipients. Am J Transpl. https://doi.org/10.1111/ajt.15891
Guillen E, Pineiro GJ, Revuelta I, Rodriguez D, Bodro M, Moreno A, Campistol JM, Diekmann F, Ventura-Aguiar P (2020) Case report of COVID-19 in a kidney transplant recipient: does immunosuppression alter the clinical presentation? Am J Transpl. https://doi.org/10.1111/ajt.15874
Seminari E, Colaneri M, Sambo M, Gallazzi I, Di Matteo A, Silvia R, Bruno R, Force CISMPT (2020) SARS Cov2 infection in a renal transplanted patients. A case report. Am J Transpl. https://doi.org/10.1111/ajt.15902
Zhu L, Xu X, Ma K, Yang J, Guan H, Chen S, Chen Z, Chen G (2020) Successful recovery of COVID-19 pneumonia in a renal transplant recipient with long-term immunosuppression. Am J Transpl. https://doi.org/10.1111/ajt.15869
Naspro R, Da Pozzo LF (2020) Urology in the time of corona. Nat Rev Urol. https://doi.org/10.1038/s41585-020-0312-1
Rombola G, Heidempergher M, Pedrini L, Farina M, Aucella F, Messa P, Brunori G (2020) Practical indications for the prevention and management of SARS-CoV-2 in ambulatory dialysis patients: lessons from the first phase of the epidemics in Lombardy. J Nephrol 33(2):193–196. https://doi.org/10.1007/s40620-020-00727-y
Acknowledgements
We thank Teri Fields for her assistance in editing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Taverna, G., Di Francesco, S., Borroni, E.M. et al. The kidney, COVID-19, and the chemokine network: an intriguing trio. Int Urol Nephrol 53, 97–104 (2021). https://doi.org/10.1007/s11255-020-02579-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-020-02579-8