
Abstract
Calf diarrhoea is one of the major problems in cattle farming with high morbidity and mortality in herds. Two enteric viruses, bovine rotavirus (BRV) and bovine coronavirus (BCoV), are the leading cause of gastroenteritis in young calves, whereas picobirnaviruses (PBVs) are often associated with diarrhoea. In the present study, the faecal specimens of 127 diarrhoeic bovines (less than 1-month-old) were employed to investigate the infection frequencies of these three pathogens. Results indicated that frequencies of BRV and BCoV in diarrhoeic calves were 38.58% and 29.92%, respectively. The 7.08% of bovine calf samples (9 out of 127) were found to be positive for PBV genogroup I. Sequence analysis further revealed the high genetic heterogeneity within representative PBV sequences. Additionally, both PBV-BCoV (n = 2) and BCoV-BRV-PBV (n = 1) co-infections were detected in bovine calves for the first time. Consequently, our findings pointed out the highly divergent nature of PBVs without regard to exact host or territory and the occasional co-existence with other enteric agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Picobirnaviruses (PBVs) are recently emergent viruses that were first discovered in human and rat stools by polyacrylamide gel electrophoresis (PAGE) in the late 1980s (Pereira et al., 1988a, 1988b). Since then, the virus has been reported in a number of species, both with and without symptoms, including domestic and wild mammals (Takiuchi et al., 2016; Navarro et al., 2017; Malik et al., 2018), avians (Ribeiro Silva et al., 2014; Masachessi et al., 2015; Verma et al., 2015) and reptiles (Fregolente et al., 2009). PBVs have been defined as opportunistic enteric—and less likely—respiratory pathogens of animals. PBVs are mostly bi-segmented, double-stranded RNA viruses, classified under the Picobirnaviridae family (Delmas et al., 2019). Segment 1 (L gene) has an RNA structure 2.2 to 2.7 kbp in length and encodes viral capsid proteins, while the 1.2–1.9 kbp segment 2 (S gene) expresses RNA-dependent RNA polymerase (RdRp), which is crucial for genome replication (Kattoor et al., 2016; Malik et al., 2017). Recently, efforts have been made to characterise the highly divergent family Picobirnaviridae, and segment 2-based classification has been the most widely utilised approach (Knox et al., 2018). According to this method, PBVs fall into two main genogroups (GI–II), and so far, the majority of known strains have been classified into the GI genogroups (Malik et al., 2014b, 2014a, 2017).
PBVs have been reported to be involved in multiple infections, with astroviruses, adenoviruses, sapoviruses and rotaviruses identified in humans (Bhattacharya et al., 2007; Giordano et al., 2008; Vu et al., 2019), with the latter also shown to be a common pathogen in co-infections in pigs (Pongsuwanna et al., 1996) and monkeys (Wang et al., 2007). A recent study further demonstrated the co-occurrence of rotaviruses and PBVs in wild birds (Serra et al., 2020). Despite strong evidence, simultaneous PBV infections with other enteric viruses have not been definitively identified in cattle. Bovine rotaviruses have been shown to be a common pathogen in PBV infections (Buzinaro et al., 2003; Mondal et al., 2013; Malik et al., 2014b). Bovine enteric coronavirus (BCoV) is an important viral agent that has a significant impact on neonatal calf diarrhoea (Hodnik et al., 2020). Two independent studies showed that the prevalence of BCoV varied greatly (5.2–39.3%) in Turkey (Hasoksuz et al., 2005; Akgül et al., 2014). It is estimated that the co-existence of BCoV infection may trigger immune system impairment and assist in the replication of other viral agents (Niskanen et al., 2002); however, the potential contribution of BCoV to the PBV infections has remained unknown.
In this study, we retrospectively assessed the maintenance and frequency of PBV infections in diarrhoeic bovine calves. The most common aetiologic viral agents causing gastrointestinal disorders in the Bovinae subfamily—BRV and BCoV—were initially screened using versatile, molecular-based methods. Further analyses focussed on detecting PBVs and evaluating their diverse nature. For this purpose, we implemented molecular detection techniques and performed sequence characterisation and phylogenetic analyses of the sequencing data from the PBV-positive samples. Finally, co-circulation of PBVs with BRV and/or BCoV was interpreted using valid statistical methods to understand the potential interactions of these agents.
Materials and methods
Collection and preparation of samples
During 2017 and 2018, diarrhoeic faecal samples of a hundred and twenty-seven calves from 1- to 30-day-old age were collected from three Middle Anatolian provinces (Sivas, Malatya and Elazig) for this study. Each of the faecal specimens was diluted 10–20% with ice-cold PBS and centrifuged at 3000 × g for 10 min to remove coarse particles. The 250 μl of supernatants was taken from the specimen aliquots for the RNA extraction using GF-1 Viral Nucleic Acid Kit (Vivantis Technologies, Malaysia) according to the manufacturer’s instructions. RNA samples were stored at − 80 °C until the reverse transcription process.
Reverse transcription polymerase chain reaction (RT-PCR)
The cDNA syntheses were carried out in a 25 μl final volume reactions containing 4 μl of RNA extract, 10 mM deoxynucleoside triphosphate (dNTP), 2,5 μl 10 × RT buffer (50 mM Tris–HCl (pH 8.3 at 25 °C), 75 mM KCl, 3 mM MgCl2 and 10 mM DTT), 50 ng of the random hexamer, 40 U RNasin, 200 U M-MuLV Reverse-Transcriptase RNase H (Vivantis, Germany). Reverse transcription (RT) reactions were performed at 37 °C for 1 h and RT enzyme was inactivated at 70 °C for 5 min.
All of the samples subjected in this study were preliminarily screened for the existence of two major viral aetiologic agents of the diarrhoea, bovine coronavirus (BCV) and rotavirus (BRV), using modified version of the previously described one-step duplex RT-PCR assay method (Zhu et al., 2011). Pre-designated primer sets were BCoVF and BCoVR targeting partial N gene (597 bp) and BRVF and BRVR targeting partial VP6 gene (383 bp) for bovine coronavirus and rotavirus, respectively. First, PCRs were conducted on cDNA samples using 10 μl of 2 × PCR Master Mix (GeneDireX, Taiwan), 1 μl of each primer (10 μM) and 5 μl of RNase free water. After the PCR mixture was aliquoted into PCR tubes, 1 μl of cDNA template (approx. 200 ng for each sample) was added resulting in a final volume of 20 μl reaction. PCRs were implemented as follows: 94 °C for 5 min, 94 °C 50 s, 55 °C 50 s, 72 °C 60 s and 72 °C 10 min. PCR products were loaded into agarose gel (1.5%) being stained with ethidium bromide (1 μl/ml) and run at 100 V for 30 min. Gels were visualised under the UV light transilluminator (MaestroGen, Taiwan) and 597 and 383 bp bright bands were considered as positive.
Templates were further subjected to PCR reaction by using detection primer set targeting putative RdRp gene previously designed by Rosen et al. (2000), which were PicoB25/PicoB43 and PicoB23/PicoB24 for GI and GII, respectively (Rosen et al., 2000) (for primer sets, see Table 1). Briefly, 40 μl PCR mixture containing 4 μl 10 × PCR buffer, 10 mM dNTP, 10 pmol/μl of each sense/antisense primer, 5 U of Taq DNA polymerase (Vivantis, Germany) and 4 μl of each RT template was prepared. Thereafter, PCR was performed for both genogroups under the following conditions: 1 cycle at 95 °C for 3 min and 40 cycles of 94 °C for 45 s, 50 °C for 45 s and 72 °C for 1 min, followed by a final elongation step of 72 °C for 10 min. The same electrophoresis procedure was repeated for detecting PBV-positive samples. These were further used to amplify ~ 1229 bp partial putative RdRp gene sequence by using the methodology previously described (Malik et al., 2018). For this purpose, PCRs were conducted in a final volume of 50 μl by using 2X Platinum™ Hot Start PCR Master Mix (Thermo Fischer Scientific), 500 nM of each primer and 100 ng of DNA template and were performed in the following conditions: 1 cycle at 98 °C for 1 min and 40 cycles of 98 °C for 10 s, 48 °C for 25 s and 72 °C for 1 min, followed by a final elongation step of 72 °C for 5 min.
Sequencing and phylogenetic analysis
Six of the PBV-positive samples were selected for further analysis. To prepare samples for the sequencing, the DNA amplicons were separated from the gel by scalpel and purified using Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI). Bidirectional sequencing was performed twice using BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on an automated sequencer (ABI 3100; Applied Biosystems, Foster City, CA).
The raw data obtained from the sequencing were quality checked manually using MEGA X 10.1 (Stecher et al., 2020) and assembled into consensus sequences. These nucleotide and amino acid data were entered in BLASTn and BLASTp tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to retrieve the most accurate data from the GenBank. Nucleotide and amino acid diversities for the each of data queries were evaluated separately. Therefore, partial genome and predicted amino acid sequences were determined. Obtained data were deposited in GenBank (accession number: MW717579-MW717582 and OK067326).
Multiple sequence alignment and phylogenetic analyses were conducted using Geneious Prime 2021.1.1 software (Kearse et al., 2012). Nucleic acid and predicted amino acid data were aligned with publicly available sequences provided by National Center for Biotechnology Information (NCBI) using MUSCLE (Edgar, 2004). For the phylogenetic analysis, the best fit model for phylogeny was selected using JModelTest (Posada, 2008). Thereafter, phylogenetic tree was built based on 358 residues predicted amino acid sequences of partial putative RdRp gene using PhyML (Guindon et al., 2010) and bootstrapped 100 times. Statistical analysis was conducted using SPSS software v. 24. Bivariate correlations were computed by Spearman’s correlation analysis with p < 0.05 regarded as significant.
Results
The prevalence of the PRV, BCoV and PBV among diarrhoeic calves
A total of 127 diarrhoeic faecal specimens were investigated to determine the prevalence of BRV, BCoV and PBV using conventional PCR and agarose gel electrophoresis. Our findings revealed that the prevalence of BRV and BCoV in the stool samples was 38.58% (49/127) and 29.92% (38/127), respectively. In addition, BRV–BCoV co-infections occurred in 18.89% of tested bovine calves (24/127).
We utilised two sets of primers, PicoB25/PicoB43 and PicoB23/PicoB24, to detect PBV-positive samples and determine the genogroups, GI and GII, respectively (Rosen et al., 2000). The results demonstrated that 7.08% of the cattle (9/127) were positive for GI, whereas no positivity was detected in the GII genogroups. Of these nine samples, two were also BCoV positive, and one sample was positive for both BCoV and BRV. Positive samples were used for further PCR experimentation to obtain a partial putative RdRp gene sequence (~ 1229 bp), which was successfully amplified from two samples. However, only one sample obtained from the Sivas province (referred to as ‘buzagi 8’) was eligible for further analyses since it had higher coverage.
Pairwise nucleotide comparison of PBV sequencing data
Selected samples were sequenced twice using chain termination methodology. We successfully obtained 201 bp sequence data from four samples and 1074 bp datum from the buzagi 8 strain. Since the genomes are highly variable in the PBV genus, nucleotide sequences were separately queried in the BLASTn database to filter out irrelevant strains from our study. Remaining sequence data demonstrating nucleotide identity above 70% were included in the multiple sequence comparison analysis.
The sequence comparison analysis based on 201 bp nucleotide data showed that Turkish strains exhibited 66.17–99.50% nucleotide identity to each other. The highest identity among samples was detected between Malatya 9 (MW717579) and the Malatya 10 (MW717580) (99.50%), whereas considerable differences were observed in the rest of the strains (varied between 65.17 and 69.65%). The Malatya 9 and Malatya 10 strains demonstrated the highest homology to raw sewage clones from Louisiana (EU938811.1) and Maryland (EU938860.1), which was between 75.12 and 76.62%. Elazig 12 (MW717581) showed 82.09% identity to the isolate PF090203 (KU729761.1), while Sivas 31 (MW717582) was closely related to the isolate C343R (KY120175.1; 94.03%). Based on 1074 bp data, buzagi 8 (OK067326) exhibited the highest relationship to otarine PBVs, specifically isolate PF090302 (KU729763.1), and shared 65.72% nucleotide identity.
Partial putative RdRp derived amino acid sequence analysis
The 66 predicted amino acid residues were determined for each sample to evaluate the potential polymorphisms on the partial putative RdRp protein sequence. Sequence comparison based on the amino acid residues revealed that Malatya 9 and Malatya 10 were identical. Elazig 12 and Sivas 31 presented 66.67% (44 out of 66) and 71.21% (47 out of 66) amino acid similarity, respectively. Furthermore, alignment between the deduced amino acid sequences and the published sequence data demonstrated that Malatya 9 and Malatya 10 showed similarity to two clones obtained from the wastewaters of Louisiana (EU938811.1) and Maryland (EU938860.1), with 84.84% agreement (56 out of 66). Sivas 31 exhibited a significant resemblance to strain C343R (ATY68938.1), presenting 98.48% (65 out of 66) amino acid similarity. Meanwhile, Elazig 12 showed equal similarity to (MG846412.1) and clone c299374 (KY928713.1) (83.33%; 55 out of 66) (Fig. 1).

Sequence demarcation analysis based on 66 residues of the partial putative RdRP protein sequences using SDT v.1.2. tool (Muhire et al., 2014). Sequencing data were aligned by MUSCLE and the pairwise identity percentage was exhibited using a full-colour scale
The 358 amino acid protein sequences deduced from the 1074 bp sequence datum of the buzagi 8 strain (OK067326) were compared with other sequences, and strain ZLY2_ct202 (QQM99864) isolated from a Tibetan antelope showed the highest similarity (267 out of 358 residues). Furthermore, evaluation of multiple-sequence alignment revealed the exact positions of multiple amino acid substitutions in the Turkish sequences. A partial sequence of buzagi 8 was positioned between the 92nd and 453rd amino acids of the putative RdRp gene, where seven catalytic active sites (A–F) of polymerase locate according to the reference strain Hy005102 (YP_239361) (Collier et al., 2016). A, C and F motifs were mostly conserved between buzagi 8 and the consensus sequence of reference strains. However, four motifs (B, D, E and G) exhibited point mutations in their predicted residues (Fig. 2). They were located as follows: F146 in motif G; T334, M338 and M344 in motif B; 374TEV376 in motif D; and F398 and M408 in motif E. In addition, motif A (LVVCTDFSKFDQH) was also preserved among Turkish prototypes.

Comparison of the 358 amino acid residues putative RdRP sequence of buzagi 8 with consensus sequence. Sequence logo above visualises consensus sequence, which was created based on the alignment of seven reference strains: strain HY005102 (YP_239361), strain 221/04–16/ITA/2004 (YP_009241386), strain monkey/KNA/2015 (YP_009361966), strain dog/KNA/2015 (YP_009389484), strain PBV/CHK/M3841/HUN/2011 (YP_009551574), isolate HKG-PF080915 and strain PBV/roe_deer/SLO/D38-14/2014. Blue arrows indicate seven catalytic active sites of polymerase (Collier et al., 2016). Disagreements in the residues are shown in coloured grids
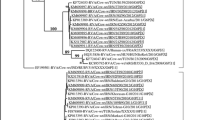
The phylogenetic tree was constructed using the alignment of 68 amino acid submissions. Phylogenetic analysis revealed that strains were segregated into four different genogroups (GI, GII, PBV-like strains and unassigned PBVs), supported by significant bootstrap values at each clade node. The majority of strains were implicated in the GI genogroup, in which buzagi 8 was clustered with a group of strains isolated from various sources, including isolate WUSTL (AVD54065) from macaques (73.22% similarity), strain ZLY2_ct202 (QMM99864) from Tibetan antelopes (74.86% similarity), isolate PBV/Human/CMRHP49B/CMR/2014 (QAA77653) from humans (72.21% similarity) and isolate Q5A/13 (AIW53314) from chickens (69.65% similarity) (see Fig. 3).

The phylogeny of picobirnavirus using deduced amino acid sequence (358 aa) of putative RdRp gene. The maximum likelihood (ML) method and the Le Gascuel model were applied to construct phylogenetic tree which was then bootstrapped one hundred times. Substitutions per site were displayed above the lines. Buzagi 8 was shown in bold letter
Discussion
Since the PBVs were detected in both diarrhoeic and clinically healthy animals, their potential role is unclear (Takiuchi et al., 2016). Furthermore, in addition to animals, PBVs have been frequently detected in invertebrates or environmental specimens (Symonds et al., 2009; Shi et al., 2016; Guajardo-Leiva et al., 2020). Thus, neither a certain host nor the mechanism of PBV transmission has been ascertained so far. Recent studies demonstrated the existence of a well-conserved prokaryotic ribosome-binding site in the viral genome (Krishnamurthy & Wang, 2018; Boros et al., 2018). In addition, mongoose and bat PBVs actually utilise mitochondrial genetic codes (Yinda et al., 2018; Kleymann et al., 2020). Based on these findings, it is posited that PBVs are viral agents of gut flora (Ghosh & Malik, 2021). This study was designed to retrospectively investigate the PBV infection frequency in diarrhoeic calves. Thus, we initially screened two major viral causative agents of diarrhoeic calves, which revealed the high incidence of rotavirus (38.58%) and coronavirus (29.92%) infection. The prevalence of rotavirus-associated diarrhoea in calves has been reported worldwide, but the results varied in Turkey (8.92–41.17%) depending on the sampling and methodology adopted (Okur Gumusova et al., 2007; Yilmaz, 2016; Aydin and Timurkan, 2018). Similarly, a recent comprehensive study reported BCoV prevalence as 21.95% in diarrhoeic calves in Turkey (Temizkan & Alkan, 2021). Taken together, our study confirmed that BCoV and BRV are dominant viral pathogens contributing to diarrhoea in young calves in Turkey.
Numerous studies have studied human and porcine PBVs worldwide (Pereira et al., 1988a; Rosen et al., 2000; Bhattacharya et al., 2006a, b; Ganesh et al., 2010; Ganesh et al., 2011a, b; Chen et al., 2014; Kylla et al., 2017). The studies focussing on PBVs of the Bovidae family were primarily conducted Brazil (Buzinaro et al., 2003; Takiuchi et al., 2016; Navarro et al., 2018) and India (Ghosh et al., 2009a; Malik et al., 2011, 2013, 2014c). The common results of these studies suggested that the frequency of PBVs varied marginally and ranged between 0.69 and 23.4%. Furthermore, the majority of identified strains were classified into the GI genogroup, whereas two GII-classified isolates have been detected in cattle so far (Malik et al., 2014b; Woo et al., 2019). On the contrary, a recent study revealed that the 201-bp partial sequence of the putative RdRp gene datum was insufficient for genotyping since it did not reflect the nucleotide diversity of the whole genome of PBVs (Knox et al., 2018; Perez et al., 2021). Our findings indicated a 7.08% positivity rate among samples (9/127), which are related to the genogroup 1 strains, showing good agreement with previous reports. Coupled with the literary evidence, these results demonstrated the intermittent presence of PBVs in diarrhoeic cattle calves in Turkey and the similarity to the strains previously defined as GI.
The evidence of concomitant PBV infection has been demonstrated for rotaviruses, astroviruses, caliciviruses and bocaviruses (Bányai et al., 2003; Bhattacharya et al., 2006a, b; Bhattacharya et al., 2007; Giordano et al., 2008; Wilburn et al., 2017). The co-occurrence of rotaviruses with PBV infection has previously been reported in piglets (Wilburn et al., 2017), humans (Bhattacharya et al., 2006a, b) and cattle (Ghosh et al., 2009b; Malik et al., 2014b). Our results supported the possibility of both PBV–BCoV dual (n = 2) and PBV–BRV–BCoV triple infections (n = 1) in bovine calves for the first time. As PBVs are often referred to as ‘opportunistic pathogens’ (Malik et al., 2014a), epithelial damages caused by rotaviruses and/or coronaviruses might have assisted PBV replication. However, the statistical analyses showed no significant correlation between the PBV and these two pathogens (p > 0.05); therefore, we conjectured that these co-existences could be a coincidence. Nonetheless, further investigation is needed to determine whether or not an interaction exists among these viruses.
Most recent studies have focussed on the amino acid sequence-based comparison of whole segment 2 data as a validated approach (Kleymann et al., 2020; Ghosh & Malik, 2021; Huaman et al., 2021). In this study, we utilised a primer pair that is capable of amplifying ~ 67% of the S segment; however, two out of nine PBV-positive detected samples presented with a detectable band only. Considering the success of the amplification, we recommend the use of Pico23-Pico25/Pico43 primer sets (Rosen et al., 2000) for routine PBV screening, whereas the PBV1.1FP/PBV1.2RP primer pair (Malik et al., 2018) can be considered for phylogenetic analysis and classification. Once high-throughput coverage was obtained, we further sought to conduct our phylogenetic analysis using 358 aa-deduced sequences of buzagi 8 (OK067326). Phylogenetically, buzagi 8 was grouped into a single clade with various strains isolated from different hosts, with amino acid similarities ranging between 69.65 and 74.86%, supported by a good bootstrap value (90%). Likewise, strain Elazig 12 (MW717581) showed the highest nucleotide homology to the isolate PF090203 (KU729761.1) obtained from a sea lion (82.09%), whereas 83.33% of the amino acids were shared with strain RS/BR/15/1S-1 (MG846412.1) and the clone c299374 (KY928713.1), which were identified from chickens and marmots, respectively. Woo et al. (2016) found a high level of variation in the PBV genome in marine mammals and emphasised that evolutionary mechanisms of the PBV genome could bear a resemblance to those of other segmented RNA viruses (Woo et al., 2016). Similarly, immense diversity between marmot-originated PBV strains was found, and nine types of PBV assortments in the PBV genome were recently proposed (Luo et al., 2018). The point mutations and genetic reassortments contribute to the evolution of PBVs (Ganesh et al., 2014; Woo et al., 2019). Additionally, Masachessi et al. (2015) also inferred that genogroup I (GGI) PBV strains could circulate in nature without any link to exact species or locations (Masachessi et al., 2015). Overall, these results point to the likelihood that the genetic heterogenicity between Turkish samples could be due to evolutionary mechanisms rather than hosts or geographical areas.
The basic polymerase domains and motifs have been explored using site-directed mutagenesis, providing an enhanced understanding of the basic mechanisms of PBV polymerases. We compared the buzagi 8 sequence with the consensus sequence of reference strains residue by residue, according to seven pre-determined motifs, A–F (Collier et al., 2016) and detected several point mutations in four of them (B, D, E and F). In addition, 13 amino acid-length motif A was further analysed for the rest of the strains, Malatya 9, Malatya 10, Elazig 12 and Sivas 31. The comparison revealed that several point mutations existed within the motifs. Interestingly, buzagi 8 had 374TEV376 residues, which differed from the consensus sequence in motif D. This motif is known to orchestrate the attachment of nucleotide substrates and determine the efficiency and fidelity of nucleotide addition (Yang et al., 2012; Collier et al., 2016). Based on these findings, we surmised that partial mutations in motif A might alter the polymerisation capability of protein, thereby resulting in further point mutations.
A basic pairwise comparison of the sequence data revealed a significant similarity among local isolates. Malatya 9 (MW717579) and Malatya 10 (MW717580) originated from the same province and displayed the highest nucleotide identity (99.50%) to each other. Furthermore, the marked resemblance of these strains with those obtained from the raw sewage from Louisiana and Maryland (75.12–76.62%). Previous studies revealed the high prevalence (100%) of the PBV genome in water-containing excrement (Symonds et al., 2009; Guajardo-Leiva et al., 2020). PBVs are also frequently considered a significant indicator of faecal contamination in drinking water sources (Symonds et al., 2009; Lin and Ganesh, 2013); therefore, it is plausible that the water sources of the cattle could function as a reservoir for PBV infection. Further, Malik et al. (2014a) demonstrated the species-wise distribution of PBVs and raw sewage isolates made up the majority of overall data (Malik et al., 2014a). Taken together, further studies are required to investigate the origin of faecal pollution of drinking water and its potential hosts in the Middle Anatolian territory.
The Sivas 31 strain (MW717582) showed the highest homology to the C343R strain—obtained from the respiratory tract of cattle in China—at the nucleotide (94.03%) and amino acid (98.48%) levels. Previous studies revealed the existence of PBVs in the alimentary and respiratory tracts of cattle (Ghosh et al., 2009a; Navarro et al., 2018; Woo et al., 2019), pigs (Smits et al., 2011; Kylla et al., 2017) and humans (Smits et al., 2012; Ng et al., 2014). Moreover, PBVs were also detected in the plasma of horses (Li et al., 2015). Mixed infection with multiple PBV variants in a single host was reported in pigs, cattle and humans (Ganesh et al., 2011a, b; Smits et al., 2011; Chen et al., 2014). This might be explained by the hypothesis that a PBV variant could spread via the bloodstream and could infect multiple organs in the host.
Several potential shortcomings need to be considered regarding the present study. First, we examined clinically diseased calves only, although PBVs could exist in healthy animals (Malik et al., 2011). Second, molecular sequencing was not applied for genotyping BRV and BCoV samples; therefore, the data lacked detail for subtypes of BRV and BCoV. Thus, there is a need for further data collection to compare the molecular characteristics of PBVs in clinically healthy calves and to determine the possible interactions between diseases, especially for the BRV and BCoV subgenotypes.
In conclusion, this study revealed a low prevalence of PBV in diarrhoeic bovine calves in Middle Anatolian provinces. Multiple sequence analysis and phylogenetic analysis of partial genomic data further demonstrated the high level of genetic diversity of PBVs in young calves. We also reported the occurrence of co-infection with BCoV in bovine calves for the first time. Our findings may be beneficial for understanding the distribution and prevalence of a neglected pathogen worldwide.
Data availability
Sequencing data obtained from this study were deposited in GenBank with accession numbers, MW717579-MW717582 and OK067326.
References
Akgül, G., Mecitoğlu, Z., Ertürk, A., Çatık, S., Temizel, E.M., Gülyaz, V., Gülaçtı, İ., Özdemir, S., Onat, K., Şenlik, B. And Şentürk, S., 2014. Isolation of First Local Coranavirus from Cattle with Winter Dysentery in Turkey Uludağ Üniversitesi Veteriner Fakültesi Dergisi, 32, 63–70
Aydin, H. and Ti̇murkan, M.Ö., 2018. Partial sequence and phylogenetic analysis of the nucleoprotein gene of coronavirus and vp7/vp4 gene of rotavirus in calf diarrhea Ataturk Universitesi Veteriner Bilimleri Dergisi, 13, 211–218
Bányai, K., Jakab, F., Reuter, G., Bene, J., Új, M., Melegh, B. and Szucs, G., 2003. Sequence heterogeneity among human picobirnaviruses detected in a gastroenteritis outbreak Archives of Virology, 148, 2281–2291
Bhattacharya, R., Sahoo, G.C., Nayak, M.K., Ghosh, S., Dutta, P., Bhattacharya, M.K., Mitra, U., Gangopadhyay, D., Dutta, S., Niyogi, S.K., Saha, D.R., Naik, T.N., Bhattacharya, S.K. and Krishnan, T., 2006. Molecular epidemiology of human astrovirus infections in Kolkata, India Infection, Genetics and Evolution, 6, 425–435
Bhattacharya, R., Sahoo, G.C., Nayak, M.K., Saha, D.R., Sur, D., Naik, T.N., Bhattacharya, S.K. and Krishnan, T., 2006. Molecular epidemiology of human picobirnaviruses among children of a slum community in Kolkata, India Infection, Genetics and Evolution, 6, 453–458
Bhattacharya, R., Sahoo, G.C., Nayak, M.K., Rajendran, K., Dutta, P., Mitra, U., Bhattacharya, M.K., Naik, T.N., Bhattacharya, S.K. and Krishnan, T., 2007. Detection of Genogroup I and II human picobirnaviruses showing small genomic RNA profile causing acute watery diarrhoea among children in Kolkata, India Infection, Genetics and Evolution, 7, 229–238
Boros, Á., Polgár, B., Pankovics, P., Fenyvesi, H., Engelmann, P., Phan, T. G., Delwart, E., & Reuter, G. (2018). Multiple divergent picobirnaviruses with functional prokaryotic Shine-Dalgarno ribosome binding sites present in cloacal sample of a diarrheic chicken, Virology, 525, 62–72.
Buzinaro, M.G., Freitas, P.P.S., Kisiellius, J.J., Ueda, M. and Jerez, J.A., 2003. Identification of a bisegmented double-stranded RNA virus (picobirnavirus) in calf faeces Veterinary Journal, 166, 185–187
Chen, M., Sun, H., Lan, D., Hua, X., Cui, L., Yuan, C. and Yang, Z., 2014. Molecular detection of genogroup I and II picobirnaviruses in pigs in China Virus Genes, 48, 553–556
Collier, A.M., Lyytinen, O.L., Guo, Y.R., Toh, Y., Poranen, M.M., Tao, Y.J., 2016. Initiation of RNA Polymerization and Polymerase Encapsidation by a Small dsRNA Virus. PLoS Pathog 12(4): e1005523.
Delmas, B., Attoui, H., Ghosh, S., Malik, Y.S., Mundt, E. and Vakharia, V.N., 2019. ICTV virus taxonomy profile: Picobirnaviridae Journal of General Virology, 100, 133–134
Edgar, R.C., 2004. MUSCLE: A multiple sequence alignment method with reduced time and space complexity BMC Bioinformatics 5 1–19
Fregolente, M.C.D., de Castro-Dias, E., Martins, S.S., Spilki, F.R., Allegretti, S.M. and Gatti, M.S.V., 2009. Molecular characterization of picobirnaviruses from new hosts Virus Research, 143, 134–136
Ganesh, B., Nataraju, S.M., Rajendran, K., Ramamurthy, T., Kanungo, S., Manna, B., Nagashima, S., Sur, D., Kobayashi, N. and Krishnan, T., 2010. Detection of closely related Picobirnaviruses among diarrhoeic children in Kolkata: Evidence of zoonoses? Infection, Genetics and Evolution, 10, 511–516
Ganesh, B., Banyai, K., Masachessi, G., Mladenova, Z., Nagashima, S., Ghosh, S., Nataraju, S., Pativada, M., Kumar, R. and Kobayashi, N., 2011. Genogroup i picobirnavirus in diarrhoeic foals: Can the horse serve as a natural reservoir for human infection? Veterinary Research, 42, 52
Ganesh, B., Nagashima, S., Ghosh, S., Nataraju, S.M., Rajendran, K., Manna, B., Ramamurthy, T., Niyogi, S.K., Kanungo, S., Sur, D., Kobayashi, N. and Krishnan, T., 2011. Detection and molecular characterization of multiple strains of picobirnavirus causing mixed infection in a diarrhoeic child: Emergence of prototype genogroup ii-like strain in Kolkata, India International Journal of Molecular Epidemiology and Genetics, 2, 61–72
Ganesh, B., Masachessi, G. and Mladenova, Z., 2014. Animal Picobirnavirus VirusDisease, 25, 223–238
Ghosh, S., & Malik, Y. S., 2021. The True Host/s of Picobirnaviruses. Frontiers in veterinary science, 7, 615293.
Ghosh, S., Kobayashi, N., Nagashima, S. and Naik, T.N., 2009a. Molecular characterization of full-length genomic segment 2 of a bovine picobirnavirus (PBV) strain: Evidence for high genetic diversity with genogroup I PBVs Journal of General Virology, 90, 2519–2524
Ghosh, S., Kobayashi, N., Nagashima, S. and Naik, T.N., 2009b. Molecular characterization of full-length genomic segment 2 of a bovine picobirnavirus (PBV) strain: Evidence for high genetic diversity with genogroup I PBVs Journal of General Virology, 90, 2519–2524
Giordano, M.O., Masachessi, G., Martinez, L.C., Barril, P.A., Ferreyra, L.J., Isa, M.B. and Nates, S. V., 2008. Two instances of large genome profile picobirnavirus occurrence in Argentinian infants with diarrhea over a 26-year period (1977-2002) Journal of Infection, 56, 371–375
Guajardo-Leiva, S., Chnaiderman, J., Gaggero, A. and Díe, B., 2020. Metagenomic insights into the sewage RNA virosphere of a large city Viruses, 12, 1–15
Guindon, S., Dufayard, J.F., Lefort, V., Anisimova, M., Hordijk, W. and Gascuel, O., 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0 Systematic Biology, 59, 307–321
Hasoksuz, M., Kayar, A., Dodurka, T. and Ilgaz, A., 2005. Detection of respiratory and enteric shedding of bovine coronaviruses in cattle in northwestern Turkey Acta Veterinaria Hungarica, 53, 137–146
Hodnik, J.J., Ježek, J. and Starič, J., 2020. Coronaviruses in cattle Tropical Animal Health and Production, 52, 2809–2816.
Huaman, J.L., Pacioni, C., Sarker, S., Doyle, M., Forsyth, D.M., Pople, A., Hampton, J.O., Carvalho, T.G., Helbig, K.J., 2021. Molecular Epidemiology and Characterization of Picobirnavirus inWild Deer and Cattle from Australia: Evidence of Genogroup I and II in the Upper Respiratory Tract. Viruses, 13, 1492.
Kattoor, J.J., Sircar, S., Saurab, S., Subramaniyan, S., Dhama, K. and Malik, Y.S., 2016. Picobirnavirus: A putative emerging threat to humans and animals Advances in Animal and Veterinary Sciences, 4, 327–331
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Meintjes, P. and Drummond, A., 2012. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data Bioinformatics, 28, 1647–1649
Kleymann, A., Becker, A., Malik, Y.S., Kobayashi, N., & Ghosh, S., 2020. Detection and Molecular Characterization of Picobirnaviruses (PBVs) in the Mongoose: Identification of a Novel PBV Using an Alternative Genetic Code. Viruses, 12(1), 99
Knox, M.A., Gedye, K.R. and Hayman, D.T.S., 2018. The challenges of analysing highly diverse picobirnavirus sequence data Viruses, 10(12): 685
Krishnamurthy, S. R., & Wang, D., 2018. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology, 516, 108–114.
Kylla, H., Dutta, T.K., Roychoudhury, P., Malik, Y.S., Mandakini, R. and Subudhi, P.K., 2017. Prevalence and molecular characterization of porcine Picobirnavirus in piglets of North East Region of India Tropical Animal Health and Production, 49, 417–422
Li, L., Giannitti, F., Low, J., Keyes, C., Ullmann, L.S., Deng, X., Aleman, M., Pesavento, P.A., Pusterla, N. and Delwart, E., 2015. Exploring the virome of diseased horses Journal of General Virology, 96, 2721–2733
Lin, J. and Ganesh, A., 2013. Water quality indicators: Bacteria, coliphages, enteric viruses International Journal of Environmental Health Research, 23, 484–506
Luo, X.L., Lu, S., Jin, D., Yang, J., Wu, S.S. and Xu, J., 2018. Marmota himalayana in the Qinghai-Tibetan plateau as a special host for bi-segmented and unsegmented picobirnaviruses article Emerging Microbes and Infections, 7, 4–11
Malik, Y.S., Chandrashekar, K.M., Sharma, K., Haq, A.A., Vaid, N., Chakravarti, S., Batra, M., Singh, R. and Pandey, A.B., 2011. Picobirnavirus detection in bovine and buffalo calves from foothills of Himalaya and Central India Tropical Animal Health and Production, 43, 1475–1478
Malik, Y.S., Kumar, N., Sharma, K., Sharma, A.K., Sircar, S., Jeena, L.M., Singh, N.K., Mondal, A., Joardar, S.N. and Balasubramanian, G., 2013. Molecular characterization of a genetically diverse bubaline picobirnavirus strain, India Thai Journal of Veterinary Medicine, 43, 609–613
Malik, Y. S., Sharma, A.K., Kumar, N., Sharma, K., Ganesh, B. and Kobayashi, N., 2014b. Identification and characterisation of a novel genogroup II picobirnavirus in a calf in India Veterinary Record, 174, 278.1-278
Malik, Y. S., Sharma, A.K., Kumar, N., Sharma, K., Ganesh, B. and Kobayashi, N., 2014c. Identification and characterisation of a novel genogroup II picobirnavirus in a Calf in India Veterinary Record, 174, 278
Malik, Y.S., Sharma, A.K., Sharma, K., Sircar, S. and Dhama, K., 2017. RNA polymerase gene based RT-PCR assay with primers update for genus specific detection of picobirnaviruses Journal of Animal and Plant Sciences, 27, 582–588
Malik, Y.S., Sircar, S., Dhama, K., Singh, R., Ghosh, S., Bányai, K., Vlasova, A.N., Nadia, T. and Singh, R.K., 2018. Molecular epidemiology and characterization of picobirnaviruses in small ruminant populations in India Infection, Genetics and Evolution, 63, 39–42
Malik, Yashpal S., Kumar, N., Sharma, K., Dhama, K., Shabbir, M.Z., Ganesh, B., Kobayashi, N. and Banyai, K., 2014a. Epidemiology, phylogeny, and evolution of emerging enteric Picobirnaviruses of animal origin and their relationship to human strains. BioMed Research International, p. 780752
Masachessi, G., Ganesh, B., Martinez, L.C., Giordano, M.O., Barril, P.A., Isa, M.B., Paván, G. V., Mateos, C.A. and Nates, S. V., 2015. Maintenance of picobirnavirus (PBV) infection in an adult orangutan (Pongo pygmaeus) and genetic diversity of excreted viral strains during a three-year period Infection, Genetics and Evolution, 29, 196–202
Mondal, A., Chakravarti, S., Majee, S.B. and Bannalikar, A.S., 2013. Detection of picobirnavirus and rotavirus in diarrhoeic faecal samples of cattle and buffalo calves in Mumbai metropolis, Western India Veterinaria Italiana, 49, 357–360
Muhire, B.M., Varsani, A., & Martin, D.P., 2014. SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PloS one, 9(9), e108277.
Navarro, R., Yibin, C., Nair, R., Peda, A., Aung, M.S., Ketzis, J., Malik, Y.S., Kobayashi, N. and Ghosh, S., 2017. Molecular characterization of complete genomic segment-2 of picobirnavirus strains detected in a cat and a dog Infection, Genetics and Evolution, 54, 200–204
Navarro, J. de O., Candido, M., de Almeida-Queiroz, S.R., Buzinaro, M. da G., Livonesi, M.C., Fernandes, A.M. and de Sousa, R.L.M., 2018. Genetic diversity of bovine Picobirnavirus, Brazil Virus Genes, 54, 724–728
Ng, T.F.F., Vega, E., Kondov, N.O., Markey, C., Deng, X., Gregoricus, N., Vinjé, J. and Delwart, E., 2014. Divergent picobirnaviruses in human feces Genome Announcements, 2, 2–3
Niskanen, R., Lindberg, A. and Tråvén, M., 2002. Failure to spread bovine virus diarrhoea virus infection from primarily infected calves Despite Concurrent Infection with Bovine Coronavirus Veterinary Journal, 163, 251–259
Okur Gumusova, S., Yazici, Z., Albayrak, H. and Meral, Y., 2007. Rotavirus and coronavirus prevalence in healthy calves and calves with diarrhoea Medycyna Weterynaryjna, 63, 62–64
Pereira, H. G., Fialho, A.M., Flewett, T.H., Teixeira, J.M.S., Andrade, Z.P., 1988a. Novel Viruses in Human Faeces. Lancet 332, 103–104
Pereira, H. G., Flewett, T.H., Candeias, J.A.N. and Barth, O.M., 1988b. A virus with a bisegmented double-stranded RNA genome in rat (Oryzomys nigripes) intestines Journal of General Virology, 69, 2749–2754
Perez, L.J., Cloherty, G.A., & Berg, M.G., 2021. Understanding the Genetic Diversity of Picobirnavirus: A Classification Update Based on Phylogenetic and Pairwise Sequence Comparison Approaches. Viruses, 13(8), 1476.
Pongsuwanna, Y., Taniguchi, K., Chiwakul, M., Urasawa, T., Wakasugi, F., Jayavasu, C. and Urasawa, S., 1996. Serological and genomic characterization of porcine rotaviruses in Thailand: Detection of a G10 porcine rotavirus Journal of Clinical Microbiology, 34, 1050–1057
Posada, D., 2008. jModelTest: Phylogenetic model averaging Molecular Biology and Evolution, 25, 1253–1256
Ribeiro Silva, R., Bezerra, D.A.M., Kaiano, J.H.L., Oliveira, D. de S., Silvestre, R.V.D., Gabbay, Y.B., Ganesh, B. and Mascarenhas, J.D.A.P., 2014. Genogroup I avian picobirnavirus detected in Brazilian broiler chickens: A molecular epidemiology study Journal of General Virology, 95, 117–122
Rosen, B.I., Fang, Z.Y., Glass, R.I. and Monroe, S.S., 2000. Cloning of human picobirnavirus genomic segments and development of an RT-PCR detection assay Virology, 277, 316–329
Serra, S., Caroline, L. and Tavares, E., 2020. Detection and molecular characterization of rotavirus and picobirnavirus in wild avians from amazon forest. Biorxiv https://doi.org/10.1101/2020.09.15.297689
Shi, M., Lin, X.D., Tian, J.H., Chen, L.J., Chen, X., Li, C.X., Qin, X.C., Li, J., Cao, J.P., Eden, J.S., Buchmann, J., Wang, W., Xu, J., Holmes, E.C., Zhang, Y.Z., 2016. Redefining the invertebrate RNA virosphere. Nature, 540(7634), 539–543.
Smits, S.L., Poon, L.L.M., van Leeuwen, M., Lau, P.N., Perera, H.K.K., Peiris, J.S.M., Simon, J.H. and Osterhaus, A.D.M.E., 2011. Genogroup I and II picobirnaviruses in respiratory tracts of pigs Emerging Infectious Diseases, 17, 2328–2330
Smits, S.L., van Leeuwen, M., Schapendonk, C.M.E., Schürch, A.C., Bodewes, R., Haagmans, B.L. and Osterhaus, A.D.M.E., 2012. Picobirnaviruses in the human respiratory tract Emerging Infectious Diseases, 18, 1539–1540
Stecher, G., Tamura, K. and Kumar, S., 2020. Molecular evolutionary genetics analysis (MEGA) for macOS Molecular Biology and Evolution, 37, 1237–1239
Symonds, E.M., Griffin, D.W. and Breitbart, M., 2009. Eukaryotic viruses in wastewater samples from the United States Applied and Environmental Microbiology, 75, 1402–1409
Takiuchi, E., Macedo, R., Kunz, A.F., Gallego, J.C., Mello, J.L. de, Otonel, R.A.A. and Alfieri, A.A., 2016. Electrophoretic RNA genomic profiles of Brazilian Picobirnavirus (PBV) strains and molecular characterization of a PBV isolated from diarrheic calf Virus Research, 211, 58–63
Temizkan, S.S and Alkan, F., 2021. Bovine Coronavirus Infections in Turkey: Molecular Analysis of the Full-Length Spike Gene Sequences of Viruses From Digestive and Respiratory Infections. Archives of Virology, 166, 2461–2468
Verma, H., Mor, S.K., Erber, J. and Goyal, S.M., 2015. Prevalence and complete genome characterization of turkey picobirnaviruses Infection, Genetics and Evolution, 30, 134–139
Vu, D.L., Sabrià, A., Aregall, N., Michl, K., Garrido, V.R., Goterris, L., Bosch, A., Pintó, R.M. and Guix, S., 2019. Novel human astroviruses: Prevalence and association with common enteric viruses in undiagnosed gastroenteritis cases in Spain Viruses, 11, 1–11
Wang, Y., Tu, X., Humphrey, C., Mcclure, H., Jiang, X., Qin, C., Glass, R.I. and Jiang, B., 2007. Detection of viral agents in fecal specimens of monkeys with diarrhea Journal of Medical Primatology, 36, 101–107
Wilburn, L., Yodmeeklin, A., Kochjan, P., Saikruang, W., Kumthip, K., Khamrin, P. and Maneekarn, N., 2017. Molecular detection and characterization of picobirnaviruses in piglets with diarrhea in Thailand Archives of Virology, 162, 1061–1066
Woo, P.C.Y., Teng, J.L.L., Bai, R., Wong, A.Y.P., Martelli, P., Hui, S.W., Tsang, A.K.L., Lau, C.C.Y., Ahmed, S.S., Yip, C.C.Y., Choi, G.K.Y., Li, K.S.M., Lam, C.S.F., Lau, S.K.P. and Yuen, K.Y., 2016. High diversity of genogroup I picobirnaviruses in mammals Frontiers in Microbiology, 7, 1–12
Woo, P.C.Y., Teng, J.L.L., Bai, R., Tang, Y., Wong, A.Y.P., Li, K.S.M., Lam, C.S.F., Fan, R.Y.Y., Lau, S.K.P. and Yuen, K.Y., 2019. Novel picobirnaviruses in respiratory and alimentary tracts of cattle and monkeys with large intra-and inter-host diversity Viruses, 11(6): 574.
Yang, X., Smidansky, E.D., Maksimchuk, K.R., Lum, D., Welch, J.L., Arnold, J.J., Cameron, C.E., & Boehr, D.D., 2012. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure, 20(9), 1519–1527.
Yilmaz, V., 2016. Investigation of Rotavirus Infection in Calves with Diarrhea in Northeast Turkey Animal and Veterinary Sciences, 4, 1
Yinda, C.K., Ghogomu, S.M., Conceição-Neto, N., Beller, L., Deboutte, W., Vanhulle, E., Maes, P., Van Ranst, M., & Matthijnssens, J., 2018. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus evolution, 4(1), vey008.
Zhu, W., Dong, J., Haga, T., Goto, Y. and Sueyoshi, M., 2011. Rapid and sensitive detection of bovine coronavirus and group A bovine rotavirus from fecal samples by using one-step duplex RT-PCR assay Journal of Veterinary Medical Science, 73, 531–534
Funding
This study was supported by the Sivas Cumhuriyet University Scientific Project Foundation (CUBAP) project no VET-029.
Author information
Authors and Affiliations
Contributions
TT and HI conceived and designed research. MOA and TT conducted all experiments. MOA and HI acquired, analysed and interpreted data. TT wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human or animal subjects performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Atasoy, M.O., Isidan, H. & Turan, T. Genetic diversity, frequency and concurrent infections of picobirnaviruses in diarrhoeic calves in Turkey. Trop Anim Health Prod 54, 127 (2022). https://doi.org/10.1007/s11250-022-03128-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11250-022-03128-4