
Abstract
This study focused on the development of vanadium-based catalysts for formic acid production from glucose. The influence of different vanadium precursors on the catalytic activity of titania supported catalysts was contemplated and compared to the performance of commercial and synthesized unsupported V2O5. The obtained results reveal a successful deposition of multiple vanadium species on TiO2 as confirmed by XRD, Raman, and UV-Vis measurements. Catalyst screening identifies V5+ species as main player indicating its important oxidizing potential. Afterwards, the key reaction conditions, as temperature, time, pressure and catalyst loading, were optimized as well as the state of the catalyst after the reaction characterized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The relentless growth in global energy demand is one of the most pressing challenges of our time. Throughout history, fossil fuels have traditionally dominated the energy landscape, constituting over 80% of primary energy sources. Oil, coal, and natural gas have been the driving forces behind industries, transportation networks and households, fostering economic growth [1, 2]. However, this reliance on fossil fuels is tightly related to concerns about resource depletion and exacerbated environmental issues as greenhouse gas emissions and consequent climate change. As the finite fossil fuel reserves are being consumed at such an accelerated rate, there is an urgent need of fundamental shift in the energy paradigm.
This backdrop sets the stage for the increasing role of hydrogen for the sustainable energy future. Hydrogen as a versatile and clean energy carrier could enable the integration of renewable energy sources but still the challenges related to its storage and transportation remain difficult to overcome. These challenges motivated the search for innovative solutions [3] and one of them is the use of liquid organic hydrogen carriers (LOHCs), providing a promising avenue to overcome the technical hurdles of hydrogen storage and transportation [4, 5]. Among the LOHCs formic acid stands out as it can be produced directly from biomass through selective catalytic oxidation [6,7,8].
The catalytic production of formic acid from biomass has been extensively reported with numerous catalytic systems [9,10,11], being vanadium-based systems the most reported because of its important catalytic activity [9, 12,13,14,15]. However, most vanadium-based catalysts operate in homogeneous phase, which complicates their separation and recyclability. Therefore, some efforts have been made to discover cost-effective, easily separable, and reusable heterogeneous catalysts [16,17,18]. Nevertheless, there is still lack of information on the conditions that the heterogeneous catalyst equals the homogeneous catalysts performance.
Therefore, the present work focuses on the study of reaction conditions that directly affect the catalytic performance of the vanadium-based systems, such as temperature, reaction time, pressure, and catalyst loading. We used a heterogeneous catalyst based on vanadium oxide supported on titania and glucose as a model biomass molecule. The results of this study not only provide information for optimizing reactions in similar systems but also present evidence of a promising alternative to the widely used homogeneous catalysts for biomass conversion into formic acid.
2 Experimental
2.1 Materials
All reagents used were of analytical grade or higher quality, and all aqueous solutions were prepared using deionized water. Commercially available support TiO2 P25 (Evonik) was stirred in water for 30 min, filtered, washed abundantly, and calcined at 450 °C for 5 h prior to use. The V2O5/TiO2 catalysts were prepared via wet impregnation, where powder titania was first stirred for 30 min with aqueous solutions of hydrated VOSO4 (Sigma-Aldrich) or NH4VO3 (Merck), evaporated at reduced pressure in a rotavapor, dried at 80 °C overnight and calcined at 450 °C for 16 h or 4 h according to the precursor used. Catalysts were prepared to obtain a nominal value of 5% w/w V and were labelled V1 for the catalyst prepared from VOSO4 and V2 for the one using NH4VO3. Additionally, the commercial VOSO4 used as precursor was also tested as a homogeneous catalyst. The commercially obtained V2O5 from Merck (V2O5 com) was used as received, while another sample was synthesized (V2O5 synt), both of which were considered for comparison purposes. V2O5 synt was prepared following a previously reported procedure [19], involving impregnation of cotton fibers with a solution containing NH4VO3 and HCl. Subsequently, these impregnated fibers were dried at 80ºC for 24 h and then calcined at 500ºC for 5 h.
2.2 Characterization
The samples were characterized by XRD (PANalytical X´Pert Pro) using Cu Kα radiation (λ = 1,5404 Å) at 45 kV and cathode current of 40 mA. Data were recorded at a step size of 0.05°and a step time of 300 s in 20–90º 2θ range.
The study of textural properties involved N2 adsorption-desorption measurements conducted at the temperature of liquid nitrogen using a Micromeritics Tristar II instrument. Prior to the analysis, the samples underwent a 2-hour degassing process at 250 ºC under vacuum conditions.
Raman spectra were recorded using a LabRAM Horiba Jobin Yvon dispersive microscope equipped with a 100 μm confocal aperture, a green laser source (λ = 532.14 nm) emitting 20 mW of power, and a CCD detector. A 50x objective lens was employed, along with D0.6 intensity filters, resulting in a laser power reduction to one-fourth of its maximum value to 5 mW. The theoretical values of the laser spot diameter and spatial resolution were 865 and 433 nm, respectively. Four scans (from 100 to 1800 cm− 1) were performed, each with an exposure time of 10 s.
Ultraviolet–visible spectroscopy (UV–Vis) was carried out using a commercial AVASPEC optical spectrometer (Avantes) equipped with an optical fiber solid sensor for wavelengths from 100 to 1000 nm with a DH-2000 lamp and a CCD detector. Barium sulfate was used as the reference for analysis. The spectra were recorded by averaging three measurements with an integration time of 150 ms.
The concentration of V in liquid phase was determined after reaction by ICP-MS using the iCAP 7200 ICP-OES Duo spectrometer from ThermoFisher Scientific.
2.3 Catalytic Activity
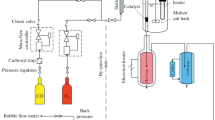
Glucose oxidation was carried out in a 50 mL stainless steel Parr reactor equipped with controllers for stirring, temperature and pressure. In a typical procedure, an aqueous solution (400 mg glucose and 20 ml water) and the catalyst (200 mg) were introduced into a PTFE liner, which was then carefully placed inside the reactor. The sealed reactor underwent one round of purging with 3.0 MPa of oxygen, followed by pressurization (from 2.0 to 3.0 MPa) and heating to reach the desired temperature (from 100 to 170 ºC). Stirring was maintained at 400 rpm throughout the process. Once the specified reaction time was reached, the reactor was transferred to an ice bath to cool to room temperature before depressurization. Subsequently, the resulting reaction product was subjected to microfiltration, dilution, and analysis using an HPLC Agilent 1260 Infinity II liquid chromatograph system. This analysis employed a Hi-Plex H column at 40.0ºC and an RI G7162A detector with a mobile phase consisting of 0.005 M H2SO4.
Fructose, arabinose, formic acid, lactic acid, and acetic acid were detected in the liquid phase, while CO2 was detected in the gas phase. The results were expressed in terms of glucose (GLC) conversion, product yield, and carbon balance, according to the following equations:
Where X could be formic, lactic, or acetic acid and C moles is referred to carbon moles.
The carbon balance considers only the products detected in liquid phase. To account for variability, a 2% error bar was applied to all reported yield results, based on the standard deviation obtained from three separate experiments.
3 Results and Discussion
The X-ray diffraction patterns of the samples are shown in Fig. 1A. The two commercial samples, V2O5 (labelled as V2O5 comm) and hydrated VOSO4 (used also as a precursor for the V1 synthesis) exhibit X-ray diffraction lines of the corresponding pure phases according to ICDD patterns 00-041-1426 and 00-001-0323, respectively. The synthesized V2O5 (labelled as V2O5 synt) also repeats the diffractions of the commercial sample without formation of any additional phase. For the supported samples V1 and V2 the diffractions corresponding to V2O5 and to TiO2 are expected. The crystalline phases of anatase and rutile, observed for the parent TiO2 P25, remain after vanadium impregnation and are clearly observed for V1 and V2 catalysts. In addition to titania diffractions, V1 and V2 catalysts present a distinct diffraction at approximately 20.3 2θ, corresponding to the most intense diffraction of bulk V2O5. However, the low intensity of this peak and the absence of all other V2O5 diffractions may indicate the formation of crystallites with dimensions below 5 nm and the presence of highly dispersed vanadium oxide species (Fig. 1B).

XRD diffractograms (A) Catalysts and support, (B) Zoom on the most intense V2O5 diffraction
The textural properties (BET surface area, pore size, and volume) of V1 and its support are summarized in Table 1. The isotherms for both samples, V1 and support, belong to type IV according to the IUPAC classification, indicating mesoporous solids. As expected after impregnation, the BET surface area, pore size, and volume become lower for the supported vanadium.
Raman spectra of V1 and corresponding support are illustrated in Fig. 2. Both samples present intense bands at approximately 152, 200, 400, 520, and 641 cm− 1 (Fig. 2A), attributed to the Raman vibrational modes of anatase. A small rutile band can be also detected at 449 cm− 1 [20, 21]. The observed baseline deviation for V1 at higher wavenumbers is attributed to fluorescence interference, associated with the formation of bulk V2O5 crystallites. As the support band dominates the spectra, we decided to subtract the spectra in order to reveal better the new bands that could be attributed to different vanadium species (Fig. 2B). Clearly, a band appeared at 998 cm− 1 with a small shoulder at 1030 cm− 1 (Fig. 2C). These bands can be related to the V = O bonds of crystalline V2O5 and monomeric VOx species, respectively. Moreover, the identification of bands at 287, 305, and 486 cm− 1, as illustrated in Fig. 2D, offers significant structural insights into the V2O5 phase, suggesting the likely formation of α-V2O5 [22, 23]. The α-V2O5 structure consists of paired V2O5 chains running parallel to each other. Within each segment of the chain, two V = O(1) vanadyl bonds run in parallel, interconnected by a V-O(3)-V bridge, while V-O(2)-V bridges act as connectors between these segments. The 704 cm− 1 band is attributed to the asymmetric stretching of V-O(2)-V bridges, whereas the symmetric stretching of the V-O(3)-V bridging bond corresponds to the vibration observed at 483 cm− 1. Raman bands below 400 cm− 1 are indicative of modes associated with angle-bending vibrations [24,25,26].

(A) General Raman spectra of the support and V1. (B) after support subtraction. (C) and (D) Zooms of the most relevant vanadium bands
The vanadium species distribution was also studied in oxygen environment using UV-Vis. The sample was gradually heated in oxygen flow from room temperature to 400ºC. As the intense charge transfer band of titania overlaps with those of vanadia, subtractions of the support’ spectra (treated in the same conditions of oxygen flow, temperature and time) were made resulting in the pure contribution of vanadium species of the sample at different temperatures (Fig. 3A). An important charge transfer band is now clearly observed at around 411 nm and corresponds to the vanadium species in square pyramidal coordination consisting in four equatorial bonds of similar length and one axial V = O bond. Another less intense band is also visible around 260 nm and corresponds to a distorted octahedral environment of vanadium. Both bands are typical of V5+ containing compounds like V2O5 [27]. A subtle contribution due to the low intense d-d transitions observed in the 550–650 nm range suggests the presence of small amount of V4+ [27]. To observe the changes in the bands produced with the temperature, an additional subtraction was made, from the initial spectra at RT all the other are subtracted (Fig. 3B). The progressive removal of water from the sample provokes the intensity increase and blue shift of the initial band at 411 nm to 409 nm. On the other hand, the gradual increase of the temperature causes the appearance of a band around 485 nm attributed to the presence of conjugated octahedral sites and V-O-V bond and V4+ d-d transitions intensification. Both bands disappear at 400ºC due to V4+◊V5+ oxidation and polymerized species dissociation to form V-O-Ti instead of V-O-V [28]. At the reaction temperature, considering that it will not exceed 200 ºC and that the continuous phase is water, we expect the presence of both V5+ and V4+ even in the presence of oxygen.

UV-VIS spectra of V1 sample at different temperature in presence of oxygen (A) vanadium species contribution. (B) vanadium species changes
3.1 Catalyst Screening and Reaction Conditions Optimization
Commercial and prepared vanadium catalysts were tested to compare the influence of various vanadium sources on formic acid production from glucose (Fig. 4). Full conversion is achieved under the catalytic conditions employed, thereby resulting in nearly equivalent yield and liquid phase selectivity. With complete conversion, the yield of formic acid over the catalysts depended on the total selectivity of formic acid. Among all catalysts tested, formic acid emerged as the major product in the liquid phase, accompanied by the generation of acetic and lactic acid as byproducts (below 2% yield). Remarkably, no significant differences were observed in the product distribution across the different catalysts. Higher performance is achieved using vanadyl sulfate as source of vanadium in comparison to ammonia metavanadate (Fig. 4A). Additionally, V2O5, commercial and synthesized, were tested and compared with a homogeneous vanadyl sulfate (Fig. 4B). The higher catalytic activity exhibited by V2O5 suggests that the V5+ species are more active than the VO2+ (V4+) ions acting as homogeneous catalyst. This is consistent with the reduction potentials found for VO2+ ◊ VO2+ (0.991 V) and VO2+ ◊ V3+ (0.337 V) [29], implying that V5+ is expected to be a better oxidizing agent.
In general, the total carbon balance is low due to the formation of gaseous CO2 during the reaction (confirmed but not quantified in this study) and reflects only the changes observed in liquid phase products distribution.

Screening of the supported vanadium oxides synthesized from different precursors (A) and unsupported vanadium catalysts (B). Reaction conditions: 400 mg glucose, 10 mg of vanadium, 20 mL of deionized water, reaction time of 150 min, 3MPa O2 pressure, and stirring speed of 400 rpm
The values obtained with V2O5 were comparable to those obtained with V1 allowing the selection of the latter as a model catalyst to study the reaction conditions.
3.2 Effect of Temperature
The influence of reaction temperature on catalytic conversion of glucose was studied in the range of 100–170 °C while keeping all the other reaction parameters constant (Fig. 5). With the increase of reaction temperature from 100 °C to 150 °C, the production of formic acid increases reaching a maximum of 40% FA yield and remains stable afterwards no matter the temperature. It is worth noting that the upper temperature limit of 170 °C was imposed owing to constraints related to the reactor liner material. One could expect a decrease in formic acid yield at higher temperatures due to the generation of more byproducts and humins [28].
The carbon balance, calculated to determine the percentage of carbon retained after the reaction, unequivocally revealed a carbon loss in these experiments. This loss as indicated above can be ascribed to the complete oxidation of the reactant and/or byproducts in CO2. The higher the conversion values, the lower the carbon balance. It is important to emphasize that at 100 °C, the lower conversion rate results in minimal over-oxidation, leading consequently to limited CO2 formation and complete carbon balance.

Effect of the reaction temperature on the conversion of glucose to formic acid. Reaction conditions: ratio of 1:2 catalyst-to-substrate, 20 mL of deionized water, reaction time of 150 min, 3MPa O2 pressure, and stirring speed of 400 rpm
3.3 Effect of Time
The effect of the reaction time on formic acid yield was also studied as presented in Fig. 6. Reaction time was defined as the duration from the moment in which the reactor temperature reaches the selected temperature of the reaction and the initiation of cooling. At time zero, corresponding to the heating process only, a low glucose conversion was observed, with a formic acid yield of 11%. Several intermediates were also detected like fructose from glucose isomerization, lactic acid, or arabinose, derived from C1-C2 cleavage and oxidation [30]. However, at this reaction time acetic acid was not found as byproduct. [31].

Effect of the reaction time on the conversion of glucose to formic acid. Reaction conditions: catalyst-to-substrate ratio 1:2, 20 mL of deionized water, reaction temperature of 150 ºC, 3MPa O2 pressure, and stirring speed of 400 rpm
After 60 min of reaction, formic acid becomes the predominant product in the liquid phase. Lactic and acetic acids appear as secondary products (less than 2% yield), more likely originated from the over-oxidation of intermediate molecules. Moreover, at that point and no matter the chosen reaction times, no significant differences in formic acid yield or product distribution are observed. The slight variations in formic acid yield are attributed more to experimental errors of handling and analysis than to a real kinetic effect.
3.4 Effect of Oxygen Pressure on Formic Acid Yield
It is widely accepted that oxygen pressure plays a key role in liquid-phase oxidation of organic compounds especially when using molecular oxygen as oxidizing agent. The oxygen pressure influences the mass transfer process involved in the movement of the oxygen from gas to the liquid phase, thereby affecting the solubility of oxygen in the reaction medium [31, 32]. The influence of starting pressure on formic acid production is shown in Fig. 7.
At 150 °C, full conversion of glucose was achieved at different pressures. Increasing the pressure from 2.0 to 2.5 MPa results in an important increase in formic acid production from 26 to 40%. However, when the pressure increases from 2.5 to 3.0 MPa, the yield remained the same, indicating that beyond 2.5 MPa, oxygen transfer was no longer the limiting step in the reaction.

Effect of employed oxygen pressure on the conversion of glucose to formic acid. Reaction conditions: catalyst-to-substrate ratio 1:2, 20 mL of deionized water, reaction temperature of 150 °C, reaction time of 150 min, and stirring speed of 400 rpm
3.5 Effect of Catalyst Loading
The oxidation of glucose by molecular oxygen on solid catalysts in aqueous solutions constitutes a three-phase process involving the gaseous phase (reactant), liquid phase (solution containing glucose), and solid phase (catalyst surface). The overall kinetics are notably contingent on the mass transfer rate [33]. In an ideal catalytic process, it is desirable to minimize the catalyst-to-glucose ratio, while simultaneously maximizing product yield, all within modest reaction conditions (including temperature, time, and pressure). For this purpose, various catalyst-to-glucose ratios were examined, and the results are shown in Fig. 8.
The reaction was conducted at 3.0 MPa pressure, thereby allowing us to dismiss the gas-to-liquid phase oxygen transfer as limiting step. Notably, the formic acid yield increases proportionally with the increase in catalyst mass up to catalyst: glucose ratio of 0.75:1, suggesting that under this ratio the limiting step occurs at the catalyst surface [34]. When the ratio increased from 0.75:1 to 1:1 insignificant modification of the formic acid yield is observed suggesting that beyond 0.75:1 no limitation on the surface occurs.

Effect of catalyst loading on the conversion of glucose to formic acid. Reaction conditions:20 mL of deionized water, reaction temperature of 150 °C, reaction time of 150 min, 3 MPa oxygen pressure, and stirring speed of 400 rpm
In summary, the reaction occurs more rapidly at high temperature (150ºC) but a loss of carbon occurs caused by reactive/intermediates over-oxidation. At this temperature short time of reaction is needed at least 2.5 MPa of oxygen pressure and catalyst/glucose ratio superior to 0.75/1 in order to avoid any possible mass transfer limitations.
The spent catalyst was characterized after the reaction (200 mg catalyst, 20 mL of deionized water, 150 min, 3.0 MPa O2 pressure, 150 °C, and 400 rpm stirring speed) in order to investigate how these reaction conditions affect the catalytic system.
XRD analysis of the spent catalyst (Fig. 9) showed no difference in anatase and rutile phases of the support before and after the reaction. However, the inset shows the disappearance of the characteristic crystalline V2O5 diffraction at 20.3 °. This can be attributed either to re-dispersion of the active phase or to a partial leaching of the active phase suggested by the yellow color obtained after the reaction. Indeed, a potential leaching of the active phase was detected in the post-reaction liquid phase, 29% of the initial vanadium content passed from the solid catalyst to the liquid phase.

XRD diffractogram of V1 before and after reaction
UV–Vis spectroscopy also shows some changes in the previously observed bands (Fig. 10A). The prominent band observed below 350 nm is primarily attributed to the ligand-to-metal charge transfer (LTCT) bands of Ti4+, overlapping with the LTCT bands of tetrahedral V5+ [35]. For enhanced clarity, Fig. 10B shows the spectra of the samples after support subtraction. We can observe a band at 420 nm, which is associated with the (O → V5+) transition. This band appears red shifted in comparison to the band indicated in Fig. 3 due to the hydration of the vanadium species during the reaction. The d-d transitions of V4+ can be also approximated at 510 and 640 nm, before and after the reaction [27]. The pre-reaction subtraction from the post reaction spectra in Fig. 10C reveals the disappearance of the bands related to V2O5, consistent with the observations made by XRD and a possible leaching phenomenon. However, it is evident that there is a higher quantity of V4+ compared to that in the catalyst before the reaction, indicated by the shape of the band in the range of 600–700 nm. This may also suggest a reduction in the oxidation state of the surface vanadium during the reaction without subsequent reoxidation.

UV-vis spectra of (A) Raw data, (B) Subtraction of support, (C) Fresh catalyst subtraction from spent catalyst
Figure 11 depicts the Raman spectra analysis of the used catalyst. To ensure reliability given the catalyst’s heterogeneous nature, we performed four distinct measurements. Notably, two distinct bands at 1281 and 1587 cm− 1 indicate the presence of coke on the catalyst’s surface, clearly visible in the magnified view in Fig. 11A [36]. Further insights into the catalyst’s alterations during the reaction were obtained by subtracting the fresh catalyst spectrum from the used catalyst spectra, presented in Fig. 11B. This subtraction highlights the disappearance of bands at 287, 305, 485, 703, and 999 cm− 1 associated with the α-V2O5 structure [22, 23], aligned with vanadium leaching as confirmed by ICP analysis. Yet, two new bands at 1026 and 1043 cm− 1 emerge, linked to monomeric vanadyl species bonded to the substrate via V-O-Ti bonds [37, 38].

Raman spectra of (A) spent catalyst V1, (B) Fresh catalyst subtraction from V1_1 spent catalyst
4 Conclusions
A series of supported and bulk vanadium-based catalyst was synthesized and used in the reaction of glucose oxidation to formic acid. In depth characterization of the supported vanadium oxide species on TiO2 support indicated the presence of V5+ and V4+ species, being the former the most active in homogeneous phase and when used as initial state during the deposition of the vanadium precursors on titania. Optimal catalytic performance was observed at 150 °C, and there were no additional changes in formic acid yield or product distribution at higher temperatures. The oxygen pressures must be situated above 2.5 MPa in order to achieve a non-limiting oxygen transfer reaction condition. The ideal catalyst-to-glucose ratio was found to be 0.75, beyond which no substantial improvement in formic acid yield appears. Post-reaction characterization suggests possible catalysts deactivation by active phase loss in a first cycle of 29%. Overall, the study underscores the significance of dispersed vanadium species in formic acid production and the sensitivity of the product yields and active phase stability to the specific conditions of use.
References
Ahmad T, Zhang D (2020) A critical review of comparative global historical energy consumption and future demand: the story told so far. Energy Rep 6:1973–1991. https://doi.org/10.1016/J.EGYR.2020.07.020
Goldemberg J (2006) The promise of clean energy. Energy Policy 34:2185–2190. https://doi.org/10.1016/J.ENPOL.2005.03.009
Li H, Cao X, Liu Y et al (2022) Safety of hydrogen storage and transportation: an overview on mechanisms, techniques, and challenges. Energy Rep 8:6258–6269. https://doi.org/10.1016/J.EGYR.2022.04.067
Salman MS, Rambhujun N, Pratthana C et al (2021) Catalysis in Liquid Organic Hydrogen Storage: recent advances, challenges, and perspectives. Ind Eng Chem Res. https://doi.org/10.1021/acs.iecr.1c03970
Southall E, Lukashuk L (2022) Hydrogen Storage and Transportation Technologies to enable the Hydrogen Economy: Liquid Organic Hydrogen Carriers: overview and perspectives on liquid organic hydrogen carriers technology. Johns Matthey Technol Rev 66:246–258. https://doi.org/10.1595/205651322X16415717819428
Kim C, Lee Y, Kim K (2023) Comparative risk assessment of a hydrogen refueling station using gaseous hydrogen and formic acid as the hydrogen carrier. Energies 16:2613. https://doi.org/10.3390/EN16062613
Zhong H, Iguchi M, Chatterjee M et al (2018) Formic acid-based Liquid Organic hydrogen Carrier System with heterogeneous catalysts. Adv Sustain Syst 2. https://doi.org/10.1002/ADSU.201700161
Eppinger J, Huang KW (2017) Formic acid as a Hydrogen energy carrier. ACS Energy Lett 2:188–195. https://doi.org/10.1021/ACSENERGYLETT.6B00574
Zhang J, Sun M, Liu X, Han Y (2014) Catalytic oxidative conversion of cellulosic biomass to formic acid and acetic acid with exceptionally high yields. Catal Today 233:77–82. https://doi.org/10.1016/J.CATTOD.2013.12.010
Wang W, Niu M, Hou Y et al (2014) Catalytic conversion of biomass-derived carbohydrates to formic acid using molecular oxygen. Green Chem 16:2614–2618. https://doi.org/10.1039/C4GC00145A
Valentini F, Kozell V, Petrucci C et al (2019) Formic acid, a biomass-derived source of energy and hydrogen for biomass upgrading. Energy Environ Sci 12:2646–2664. https://doi.org/10.1039/C9EE01747J
Achour M, Álvarez-Hernández D, Ruiz-López E et al (2023) Formic acid as renewable reagent and product in biomass upgrading. Tetrahedron Green Chem 2:100020. https://doi.org/10.1016/J.TGCHEM.2023.100020
Lu T, Hou Y, Wu W et al (2018) Catalytic oxidation of cellulose to formic acid in V(V)-Fe(III)-H2SO4 aqueous solution with O2. Fuel Process Technol 173:197–204. https://doi.org/10.1016/J.FUPROC.2018.02.001
Albert J (2017) Selective oxidation of lignocellulosic biomass to formic acid and high-grade cellulose using tailor-made polyoxometalate catalysts. Faraday Discuss 202:99–109. https://doi.org/10.1039/C7FD00047B
Gromov NV, Medvedeva TB, Lukoyanov IA et al (2023) Hydrolysis-oxidation of cellulose to formic acid in the presence of micellar vanadium-containing molybdophosphoric heteropoly acids. Results Eng 17:100913. https://doi.org/10.1016/J.RINENG.2023.100913
Wu L, Yang Y, Cheng J et al (2022) Highly efficient Conversion of carbohydrates into Formic Acid with a heterogeneous MgO Catalyst at Near-Ambient temperatures. ACS Sustain Chem Eng 10:15423–15436. https://doi.org/10.1021/acssuschemeng.2c04502
Li X, Zhang L, Wang S, Wu Y (2020) Recent advances in aqueous-phase Catalytic conversions of Biomass platform Chemicals over heterogeneous catalysts. Front Chem 7:508539. https://doi.org/10.3389/fchem.2019.00948
Li J, Smith RL, Xu S et al (2022) Manganese oxide as an alternative to vanadium-based catalysts for effective conversion of glucose to formic acid in water. Green Chem 24:315–324. https://doi.org/10.1039/D1GC03637H
Alghool S, Abd El-Halim HF, Mostafa AM (2019) An eco-friendly synthesis of V2O5 nanoparticles and their Catalytic activity for the degradation of 4-Nitrophrnol. J Inorg Organomet Polym Mater 29:1324–1330. https://doi.org/10.1007/s10904-019-01096-1
Tuschel D (2019) Raman spectroscopy and polymorphism. Spectroscopy 34:10–21
Lakshmi-Narayana A, Hussain OM, Mauger A, Julien CM (2019) Transport properties of nanostructured Li2TiO3 anode material synthesized by hydrothermal method. Sci 1:56. https://doi.org/10.3390/sci1030056
Smirnov MB, Roginskii EM, Smirnov KS et al (2018) Unraveling the structure-raman spectra relationships in V2O5 polymorphs via a comprehensive experimental and DFT study. Inorg Chem 57:9190–9204. https://doi.org/10.1021/acs.inorgchem.8b01212
Shvets P, Dikaya O, Maksimova K, Goikhman A (2019) A review of Raman spectroscopy of vanadium oxides. J Raman Spectrosc 50:1226–1244. https://doi.org/10.1002/JRS.5616
Went GT, Leu L, jen, Bell AT (1992) Quantitative structural analysis of dispersed vanadia species in TiO2(anatase)-supported V2O5. J Catal 134:479–491. https://doi.org/10.1016/0021-9517(92)90336-G
Ganjkhanlou Y, Janssens TVW, Vennestrøm PNR et al (2020) Location and activity of VOx species on TiO2 particles for NH3-SCR catalysis. Appl Catal B 278:119337. https://doi.org/10.1016/J.APCATB.2020.119337
Chai S, Li S, Li W et al (2022) Fabrication of high loading V2O5/TiO2 catalysts derived from metal-organic framework with excellent activity for chlorobenzene decomposition. Appl Surf Sci 572:151511. https://doi.org/10.1016/J.APSUSC.2021.151511
Burcham LJ, Deo G, Gao X, Wachs IE (2000) In situ IR, Raman, and UV-Vis DRS spectroscopy of supported vanadium oxide catalysts during methanol oxidation. Top Catal 11:85–100. https://doi.org/10.1023/A:1027275225668
Shan H, Li L, Bai W, Liu L (2022) Evolution process of Humins Derived from glucose. ChemistrySelect 7:e202201237. https://doi.org/10.1002/SLCT.202201237
Lide DR (2004) CRC handbook of chemistry and physics. CRC
Niu M, Hou Y, Wu W et al (2018) Successive C1–C2 bond cleavage: the mechanism of vanadium(v)-catalyzed aerobic oxidation of d -glucose to formic acid in aqueous solution. Phys Chem Chem Phys 20:17942–17951. https://doi.org/10.1039/C8CP02352B
Hobbs CC, Drew EH, Hof HAV et al (1972) Mass-transfer rate-Limitation effects in Liquid-Phase Oxidation. Industrial Eng Chem Prod Res Dev 11:220–225. https://doi.org/10.1021/i360042a020
Tromans D (1998) Temperature and pressure dependent solubility of oxygen in water: a thermodynamic analysis. Hydrometallurgy 48:327–342. https://doi.org/10.1016/S0304-386X(98)00007-3
Delidovich IV, Moroz BL, Taran OP et al (2013) Aerobic selective oxidation of glucose to gluconate catalyzed by Au/Al2O3 and Au/C: impact of the mass-transfer processes on the overall kinetics. Chem Eng J 223:921–931. https://doi.org/10.1016/J.CEJ.2012.11.073
Tschentscher R, Nijhuis TA, Van Der Schaaf J, Schouten JC (2012) Glucose oxidation in slurry reactors and rotating foam reactors. Ind Eng Chem Res 51:1620–1634. https://doi.org/10.1021/ie200694z
Sushchenko E, Kharlamova T (2016) Textural characteristics and structural peculiarities of supported vanadium oxide materials prepared by the NH4VO3 aqueous solution wetness impregnation. Key Eng Mater 670:101–106. https://doi.org/10.4028/www.scientific.net/KEM.670.101
Guerrero-Pérez MO, Bañares MA (2006) From conventional in situ to operando studies in Raman spectroscopy. Catal Today 113:48–57. https://doi.org/10.1016/J.CATTOD.2005.11.009
Went GT, Oyama ST, Bell AT (1990) Laser Raman spectroscopy of supported vanadium oxide catalysts. J Phys Chem 94:4240–4246
Schraml-Marth M, Wokaun A, Pohl M, Krauss H-L (1991) Spectroscopic investigation of the structure of silica-supported vanadium oxide catalysts at submonolayer coverages. J Chem Soc Faraday Trans 87:2635–2646
Acknowledgements
Financial support was obtained from Spanish Ministerio de Ciencia e Innovación (MCIN/AEI /https://doi.org/10.13039/501100011033/) and for FEDER Funds “una manera de hacer Europa”, Project PID2020-113809RB-C32.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Álvarez-Hernández, D., Ivanova, S., Domínguez, M.I. et al. V2O5/TiO2 Catalyst for Catalytic Glucose Oxidation to Formic Acid in Batch Reactor: Vanadium Species Nature and Reaction Conditions Optimization. Top Catal (2024). https://doi.org/10.1007/s11244-024-01982-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s11244-024-01982-0