
Abstract
The aqueous phase reforming of glycerol, to hydrogen, alkanes and liquid phase dehydration/dehydrogenation products, was studied over a series of 1 wt% Pt/LaMO3 (where M = Al, Cr, Mn, Fe, Co, Ni) catalysts and compared to a standard 1 wt% Pt/γ-Al2O3 catalyst. The sol–gel combustion synthesis of lanthanum-based perovskites LaMO3 produced pure phase perovskites with surface areas of 8–18 m2g−1. Glycerol conversions were higher than the Pt/γ-Al2O3 (10%) for several perovskite supported catalysts, with the highest being for Pt/LaNiO3 (19%). Perovskite-based catalysts showed reduced alkane formation and significantly increased lactic acid formation compared to the standard catalyst. However, most of the perovskite materials undergo phase separation to LaCO3OH and respective M site oxides with Pt particle migration. The exception being the LaCrO3 support which was found to remain structurally stable. Catalytic performance remained stable over several cycles, for catalysts M = Al, Cr and Ni, despite phase separation of some of these materials. Materials where M site leaching into solution was observed (M = Mn and Co), were found to be catalytically unstable, which was hypothesised to be due to significant loss in support surface area and uncontrolled migration of Pt to the remaining support surface. In the case of Pt/LaNiO3 alloying between the exsoluted Ni and Pt was observed post reaction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Hydrogen has the potential to replace conventional fossil fuels as a clean energy carrier, with applications in electric generation, storage and transportation, through utilisation of fuel cell technology and direct combustion in turbines and heating [1,2,3]. Current industrial production methods, such as methane steam reforming, result in depletion of finite fossil fuels and high CO2 emissions [4]. While these current production methods will remain important, a whole host of other production technologies with lower CO2 emission are required.
Aqueous phase reforming (APR) utilises waste oxygenates derived from biomass sources such as methanol, ethylene glycol, sorbitol, and glycerol for the renewable production of hydrogen. The process was first described by Dumesic et al. using operating conditions at low temperatures (200–250 °C) and intermediate pressures (15–50 bar) [5,6,7]. Thermodynamically, the low operation temperature favours the water gas shift (WGS) reaction which results in low CO concentrations in the feed and is therefore favourable for fuel cell applications [8]. Additionally, the energy demand for APR is reduced compared to steam reforming due to the lower operating temperature and no requirement to vaporise the solvent [9].
Several active metals have been utilised for APR, including Pt, Ni, Pd, Ru [10,11,12]. Pt is commonly used due to its high activity for C–C bond scission and WGS, whilst maintaining low methanation activity [13]. The choice of catalyst support material is also key to controlling the selectivity and reaction pathways in APR. Pt/γ-Al2O3 is a promising candidate for the APR reaction, displaying favourable acid–base properties, good activity, and high surface areas [14]. Yet, the material undergoes phase transition to boehmite under reaction conditions, resulting in increased surface acidity and decreased catalytic activity [15, 16]. Significant work to improve the stability of γ-Al2O3 includes the use of silica or ZnO coatings [17, 18]. Alternatively, Guo et al. evaluated the performance of a range of other support materials with different basicity and found the more basic materials facilitated WGS reaction and higher hydrogen selectivity [19]. However, these catalysts display low hydrothermal stability. Other materials such as hydrotalcites and spinels have been utilised to increase the hydrothermal stability and catalytic activity [20,21,22,23].
Perovskites are mixed metal materials with the structural formula ABO3 [24] that are known to have structural versatility and thermal stability, which has led to widespread use in a variety of applications [25]. Previous studies have shown Ni/LaAlO3 and Ni-Cu/LaAlO3 catalysts have enhanced performance and stability in the APR process, relative to Ni/γ-Al2O3 [26, 27]. Yet, the structure diversity of the perovskite, with numerous possible A and the B sites [28], has not been exploited in the APR reaction. Given the diversity in catalytic activity and selectivity of lanthanum perovskites of various B site composition, for a range of reactions including glycerol oxidation [29, 30], we have evaluated the APR performance of Pt nanoparticles supported on LaMO3 catalysts (where M = Al, Cr, Mn, Fe, Co, Ni). The catalytic performance of APR of glycerol is compared with a Pt/γ-Al2O3 standard catalysts. Analysis of the main liquid products highlighted differences between the perovskite and alumina catalysts with structure–activity relationships and catalyst stability being discussed.
2 Experimental
2.1 Catalyst Preparation
The perovskite materials were prepared by a sol–gel combustion method discussed in detail in the following reference [31]. Briefly, La(NO3)3·6H2O, M(NO3)x·yH2O, [where M = Al, Cr, Mn, Fe, Co, Ni; x = 2,3, y = 4,6] were added in stoichiometric amounts. Citric acid was then added in a 2:1 ratio to metal nitrates and dissolved in minimum amount of deionised water. The pH of the resultant solution was adjusted to 7 using 3 M ammonia solution and aged at 130 °C until gel formation. The gel was then combusted at 400 °C for 10 min and further calcined at the required temperature (Table 1), as determined by thermogravimetric analysis (TGA) (Figure S1), for 2 h.
LaCO3OH was prepared by a hydrothermal synthesis method [32]. In a standard preparation, equimolar amounts of NaOH and Na2CO3 were dissolved in 40 ml deionised water. The solution was added into a 20 ml solution of La(NO3)3·6H2O under stirring. The resultant white slurry was transferred to a 125 ml autoclave and held at 230 °C for 8 h. Once cooled, the resultant powder was washed with ethanol and deionised water and dried overnight at 90 °C.
The 1 wt% Pt supported catalysts were then prepared by a conventional wet impregnation method. The requisite amount of PtCl4 solution was dispensed and deionised water was added to give an overall solution of 16 ml. The mixture was vigorously stirred at 60 °C. The support (0.99 g) was periodically added slowly over a period of 20 min. The resulting slurry was stirred for 15 min before heating to 95 °C and left to dry overnight. The dried powder was then ground and calcined in air at 450 °C for 2 h.
2.2 Catalyst Characterisation
X-ray powder (XRD) patterns of the catalysts were recorded with a Bruker d8 discover with a monochromated Co source operating at 35 kV and 40 mA and a Vantec detector in the 2θ scan range 20–100°, using a 0.014° step size. Patterns were matched using ICDD database PDF with a list of database files is given in Table S1. N2 adsorption experiments were performed at − 196 °C (Micromeritics Tristar 3000) to obtain surface areas determined by BET method. Before measurements, the required amount of sample was measured and degassed overnight at 90 °C. Thermogravimetric analysis (TGA) of the samples was carried out using a TA SDT Q600 to investigate the formation temperature of the perovskites. The samples were heated to 900 °C under air with a 10 °C/min ramp rate. CO chemisorption measurements were performed using a Quantachrome ChemBET. 100 mg of sample was loaded into 125 μL loop in-between quartz wool and reduced under 10% H2 flow at 240 °C for 2 h and cooled to room temperature under N2 flow. For analysis, Helium carrier was used, allowing the detector to stabilise for approx. 45 min and consequently titrating with 10% CO (injection vol. 12.5 uL). Data-processing then performed on the Quantachrome software. ICP-OES experiments were performed on reaction filtrates using an Agilient 4210 MP-OES fitted with a SPS4 autosampler to determine extent of metal leaching. Pt L3-edge X-ray absorption spectra (XAS) were collected in fluorescence mode on the B18 beamline at Diamond Light Source using the Si (311) monochromator. All samples were pressed into a pellet using cellulose as a diluent. The XAS data and the extended X-ray absorption fine structure (EXAFS) were processed and fitted using IFEFFIT with the Horae package (Athena and Artemis) [33, 34].The fit range for the EXAFS data was 2.5 < k < 12.5 A−1, 1.0 < R < 2 Å (Fresh) and 1.0 < R < 3.3 Å (Used). XPS data was acquired using a Kratos Axis SUPRA using monochromated Al kα (1486.69 eV) X-rays at 15 mA emission and 12 kV HT (180 W) and a spot size/analysis area of 700 × 300 µm. High resolution spectra were obtained using a pass energy of 20 eV, step size of 0.1 eV and sweep time of 60 s. Survey spectra were obtained using a pass energy of 160 eV. Charge neutralisation was achieved using an electron flood gun with filament current = 0.38 A, charge balance = 2 V, filament bias = 4.2 V. Spectra have been charge corrected to adventitious carbon at 284.8 eV. All data was recorded at a base pressure of below 9 × 10−9 Torr and a room temperature of 294 K. Data was analysed using CasaXPS v2.3.19PR1.0. Peaks were fit with a Shirley background prior to component analysis. Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100 operating at 200 kV. Samples were prepared by dry dispersion on 300 mesh copper grids coated with holey carbon film.
2.3 Catalytic Testing
Catalyst testing was carried out using a Parr 5500 series bench top micro reactor (50 ml) equipped with a Parr 4848 reactor controller system. The catalysts were tested using a standard procedure where 20 ml of 10 wt% glycerol solution and 60 mg of the catalyst were loaded into the autoclave. The reaction was then carried out over a period of 2 h at 240 °C, 42 bar Ar, 1000 RPM. Gas products were collected and analysed using a Varian 3800 GC equipped with a TCD and FID detector and a Shincarbon ST column to give H2, CO, CO2, CH4, C2H6, and minor peaks for ethene and propene. The reactant and products in liquid phase were analysed by HPLC using a Hitachi Chromaster equipped with an Agilent Metacarb 67H column and a refractive index detector. The products observed included Lactic Acid (LA), Ethylene Glycol (EG), Hydroxyacetone (HA), 1,2-Propanediol, 2-Propanol (2-P), 1-Propanol (1-P), and Ethanol (E). Calculations were carried out as follows:
\({\text{Turnover frequency}}\;TOF \left( {h^{ - 1} } \right) = \left( {\frac{{\left( {{{\left[ {glycerol} \right]_{in} - \left[ {glycerol} \right]_{out} } \mathord{\left/ {\vphantom {{\left[ {glycerol} \right]_{in} - \left[ {glycerol} \right]_{out} } {mol\, Pt}}} \right. \kern-0pt} {mol\, Pt}}} \right)}}{time}} \right)\)
where R = stoichiometric reforming ratio of 2.33
where Ps = specified carbon product; Pi = carbon product; Cn = carbon number
3 Results and Discussion
3.1 Fresh Catalyst Characterisation
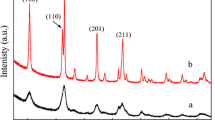
Powder X-ray diffraction (XRD) patterns of the synthesised Pt/LaMO3 and Pt/Al2O3 are given in Fig. 1. For all catalysts, no peaks associated with Pt or PtOx were found, due to the low loading (1 wt%) and probable dispersed nature of the particles. Perovskite phases are predominant in all synthesised LaMO3 materials, with clear splitting of reflections indicating the presence of the expected rhombohedral or orthorhombic phases. Minor by-product phases of LaCrO4 and NiO are observed in the LaCrO3 and LaNiO-3 catalysts respectively. The presence of LaCrO4 intermediate is consistent with previous reports [35]. These phases could be further minimised by higher calcination temperature. However, the reduction in surface area associated with higher calcination temperatures would be counterproductive for Pt dispersion.

Powder X-ray diffraction patterns of prepared a Pt/Al2O3 (black), Pt/LaAlO3 (red), Pt/LaCrO3 b Pt/LaMnO3 (green), Pt/LaFeO3 (grey), Pt/LaCoO3 (orange), Pt/LaNiO3 (pink). Phases:  perovskite;
perovskite; γ-Al2O3;
γ-Al2O3;  LaCrO4; and
LaCrO4; and  NiO
NiO
Table 1 shows the physicochemical properties of the as prepared catalysts. The calcination temperatures for perovskite were determined by thermogravimetric analysis (TGA) of the amorphous combusted gels (Figure S1). Surface area values for the perovskite materials ranged between 8 and 18 m2g−1 and are significantly lower than the 127 m2g−1 of Pt/γ-Al2O3. CO chemisorption of the fresh catalysts, prior to sufficient reduction, resulted in little CO uptake and is suggestive of the Pt being present in an oxidised form. Pre-reduction of the catalysts for 2 h at the APR reaction temperature of 240 °C resulted in a range of Pt dispersions between 42 and 77% on the perovskite supported catalysts. By comparison, a Pt dispersion of 52% was observed for Pt γ-Al2O3. The dispersions on the perovskite supports can be considered quite exceptional given their relatively low surface area. It is not anticipated that M site metals in the LaMO3 structures will have reduced to metallic form during this reduction process.
XPS determined binding energies for La, M, Pt species and surface At % of the synthesised perovskites are summarised in Table 2, and shown in Fig. 2 and 3. The observed La 3d peaks at 834–835 eV with a band gap of 3.9 eV could be assigned to La(OH)3 species and is consistent with previous reports for the oxide and perovskite materials [36, 37]. However, the band gap splitting of 3.5 eV for the LaNiO3 La 3d species at 834.17 eV suggests the presence of La2(CO3)3, possibly from carbonation in air or the presence of phase separated La species [38]. The M site 2p regions have peaks indicative of 3 + species with satellite features similar to the reported single oxides and perovskites [39,40,41,42]. The Cr 2p bands can be assigned to Cr3+ at 576.11 eV and Cr5+/Cr6+ at 579.36 eV, the intensity of the Cr5+/Cr6+ peak is consistent with the XRD pattern analysis and the presence of minor LaCrO4 phase, however, the presence of surface Cr5+/Cr6+ is also suggested as stabilisation of Cr at the surface of samples [43]. The Al 2p peaks at 73–75 eV overlaps with the Pt 4f doublets at 74.5 eV assigned as PtO2. The most intense Ni 2p peak at 855.21 eV overlaps with the La 3d3/2 peaks leading to difficulties in peak analysis, however the peaks can be assigned as Ni2+/Ni3+ and agree with previous literature with the presence of LaNiO3 phase [36, 44, 45]. The Pt 4f species gives doublets at binding energies of 74.4–75.0 eV and 72.2–73.4 eV assigned as PtO2 and PtO species respectively [46].

XPS spectra of La 3d/Ni 2p and Pt 4f/Al 2p regions for a Pt/LaNiO3, b Pt/LaCoO3, c Pt/LaFeO3, d Pt/LaMnO3, e Pt/LaCrO3, f Pt/LaAlO3, g Pt/Al2O3

XPS Spectra of M 2p region for Pt/LaCrO3, Pt/LaMnO3, Pt/LaCoO3, Pt/LaFeO3
TEM micrographs of the fresh Pt/LaMO3 materials show similar structural behaviour, with minor differences between each catalyst (representative images shown in Fig. 4). Due to small clusters and oxide form of Pt, it was difficult to analyse the Pt particle size for Pt/LaMO3 (where M = Al, Co, Ni), with Pt not seen even at high magnification (Figure S2). Particle sizes that could be determined by TEM (Table 1) correspond with the chemisorption results, showing highly dispersed small particles of PtOx. The slightly higher particle sizes seen by TEM vs chemisorption being accounted by the limited resolution of the instrument not accounting for sub-nanometre particles and also the limited number of particles counted by TEM.

TEM micrographs of freshly prepared a Pt/LaAlO3 and b Pt/LaCrO3
Finally, Pt L3-edge XAS of the fresh catalysts was performed to elucidate oxidation state and provide information on structure. Linear combination fitting of the XANES showed, in agreement with XPS, that the bulk structure in all fresh samples comprised of PtO2 with trace Pt(0) seen in Pt/γ-Al2O3 (16 at.%). The magnitude of the EXAFS Fourier Transform of all catalysts (Figure S3–S9) showed a reduced signal associated with Pt–Pt distances relative to an PtO2 standard (Fig. 5). The extent of this signal reduction prevented accurate fitting of this path with only the first shell Pt–O distance being fitted. As anticipated for metal oxides, the coordination number for 1st shell Pt–O in all samples was, within error 6, that of bulk PtO2. Although, the Pt–Pt coordination couldn’t be fitted, the data can be used qualitatively to show that the long-range order of PtO2 is significantly reduced in the catalysts compared to bulk PtO2, due to the highly dispersed nature of particles. In summary, all LaMO3 perovskites of reasonable phase purity were prepared and successfully supported highly dispersed PtOx species, which were reducible at 240 °C to give highly dispersed Pt.

Pt L3 edge EXAFS Magnitude of the Fourier Transform χR of PtO2 (black), PtLaCrO3 Fresh (red) and PtLaMnO3 Fresh (blue)
3.2 Catalytic Performance in the APR of Glycerol
The catalyst performance of the perovskite-based catalysts was investigated for the APR reaction of 10 wt% aqueous pure glycerol, in a batch reactor and under optimised conditions (240 °C, 1000 RPM, 42 bar) as determined by Subramanian et al. [14]. The catalyst performance of the perovskite and standard catalysts over a 2 h reaction is shown in Table 3. Prior to discussion of the catalytic results, it is important to note that the support materials show no activity for the APR reaction, confirming the importance of Pt.
Catalytic performance of the 1 wt% Pt/γ-Al2O3 standard was found to be comparable with results reported by Subramanian et al., under identical reaction conditions using a 2 wt% Pt/γ-Al2O3 catalyst, with TOFs of 346 h−1 and 368 h−1 respectively. Hydrogen production rates are also comparable, with formation rates 260 vs 371 µmol−1min−1gcat−1. The slightly lower TOF seen for the catalyst within this study can be attributed to a lower substrate/metal molar ratio [14]. Further, a H2:CO2 ratio of 1.98 was observed in the current study, which is lower than the 2.8 reported by Subramanian et al. Values below the stoichiometry value of 2.33 were previously observed by Wawrzetz et al., with a H2:CO2 ratio of 1.90 and were attributed to hydrogen consumption in the hydrogenation of unsaturated intermediates [47]. Therefore, it can be concluded that similar hydrogen consuming reaction pathways are seen in the present APR reaction.
Activity towards glycerol conversion, expressed as percentage conversion or TOF, for the perovskite supported Pt was found to vary from 6 to 19%/205–652 h−1. Normalisation of TOF (Table S2) by Pt dispersion showed little correlation between activity and Pt surface area. The conversion for glycerol was highest for Pt/LaNiO3 (19%), which along with Pt/LaAlO3 and Pt/LaCrO3 was higher than the Pt/γ-Al2O3 standard. While conversion was higher for these perovskite supported catalysts, conversion to gas and hydrogen selectivity (calculated from the H2:CO2 ratio of reforming glycerol) was lower than the Pt/γ-Al2O3 standard. Consequently, hydrogen production rates were only modestly higher than for the standard, with the highest being 282 µmol−1min−1g −1cat for Pt/LaNiO3 compared to 260 µmol−1min−1g −1cat for Pt/γ-Al2O3. Lower conversion of glycerol was exhibited by Pt/LaMnO3, Pt/LaFeO3, and Pt/LaCoO3 catalysts (lowest for Pt/LaFeO3 at 6%). Interestingly, aside from Pt/LaFeO3, which exhibited a low H2 formation rate, the higher conversion to gas and hydrogen selectivities resulted in hydrogen production rates not dissimilar to the higher conversion perovskite supported catalysts. Despite low conversion, the Pt/LaCoO3 catalyst exhibited the highest conversion to gas (19%), hydrogen formation rate (331.8 μmolmin−1g −1cat ) and hydrogen selectivity (75%).
Notably, the H2: CO2 ratios are significantly above the stoichiometry value for nearly all the perovskite catalysts, aside from Pt/LaNiO3, which indicates hydrogen production is occurring from competing reactions rather than the direct reforming reaction. The higher hydrogen formation rates coupled with low conversion to gas for the perovskite-based catalysts also supports reduced reforming activity when compared to competing reactions.
3.3 Carbon Product Analysis
The carbon selectivity for a range of liquid and gas phase carbon products is shown in Fig. 6. Pt/γ-Al2O3 and Pt/LaAlO3 showed similar carbon product selectivity, with modestly decreased alkane and CO2 selectivity in the latter and increased lactic acid at the expense of hydroxyacetone. Proposed reaction pathways postulate hydroxyacetone as a first intermediate from glycerol dehydration, which can then convert to lactic acid [47, 48]. Pt/LaCrO3 and Pt/LaNiO3 also showed comparable selectivity profiles, with subtle differences in 1,2 propanediol and hydroxyacetone selectivities. The low conversion, but high hydrogen yielding reactions with Pt/LaMnO3 and Pt/LaCoO3, showed very high lactic acid selectivity and very low CO2 selectivity, supporting the prior hypothesis that dehydration and dehydrogenation reaction pathways predominate over C–C scission (although ethanol is observed in comparable selectivity in all reactions). The prevalence of these reaction pathways could be due to the acid–base character of the perovskite materials as it has been reported that lactic acid formation occurs under basic hydrothermal conditions [49,50,51].

Carbon product selectivity for APR of glycerol over Pt/LaMO3 and Pt/Al2O3. Products: Lactic Acid (LA), Ethylene Glycol (EG), Hydroxyacetone (HA), 1,2-Propanediol, 2-Propanol (2-P), 1-Propanol (1-P), and Ethanol (E). Reaction conditions: 240 °C, PAr = 42 bar, 1000 RPM, 20 ml 10 wt% glycerol, 60 mg catalyst, 2 h. Error is the standard deviation of these tests
The perovskite catalysts also showed lower formation of alkanes than Pt/γ-Al2O3, aside from Pt/LaNiO3, which can be attributed to the potential exposed Ni(0) sites formed under reaction that are known to have higher intrinsic methanation rates [52]. CO was undetected in almost all the reaction analysis suggesting high WGS activity, although the Pt/LaNiO3 catalyst showed a small amount of CO, possibly due to the higher conversion or reduced WGS activity through Ni defect sites.
3.4 Catalyst Re-use
Repeat reactions were carried out for several catalysts, under identical reaction conditions, to elucidated catalyst deactivation (Fig. 7) as often seen in the APR of glycerol [7]. To note; Pt/LaFeO3 was not tested due to poor initial catalytic performance and Pt/LaCoO3 was not tested due to evident leaching of Co into the reaction solution (i.e., visible colour change of solution post reaction). Glycerol conversions remained stable after multiple reuses for each of the tested catalysts, apart from Pt/LaMnO3which saw conversion dramatically dropped after a single catalytic run. Pt/γ-Al2O3 showed an initial slight increase in activity after the first run, which has been previously observed [14, 48]. However, it is known that upon prolonged testing this catalyst deactivates, due to the phase change to boehmite, the presence of glycerol or carbonaceous product on surface blocking active sites, or Pt sintering [48]. Interestingly, the rates of hydrogen production and hence hydrogen selectivity for nearly all catalysts significantly reduces despite stable conversions indicating changes in reaction pathways possibly from structural changes or site blockage.

a Glycerol conversion and b) Hydrogen formation rate for Pt/Al2O3 and Pt/LaMO3 (where M = Al, Cr, Mn, Ni) over several cycles. Reaction conditions: 240 °C, PAr = 42 bar, 1000 RPM, 20 ml 10 wt% glycerol, 60 mg catalyst, 2 h. Carbon mass balance for all reactions is between 95 and 104%
3.5 Post Reaction Catalyst Characterisation
MP-AES analysis of the reaction effluents, shown in Table 4, showed none or limited leaching of La or Pt during rection. Leaching of M sites was seen for nearly all perovskite catalysts; however, the extent of leaching was different for each material. The highest degree of leaching was observed for Co and Mn perovskites, while Cr leaching was undetectable. Co leaching from perovskites is in agreement with X. Yang et al. who tested LaCoO3 catalysts for lactic acid production from cellulosic biomass and noted some Co leaching, however this was notably less extreme than seen currently [53]. The presence of Al leaching in LaAlO3 was also surprising given the reported stability of this support during APR [26, 27].
The XRD patterns of the catalysts post-reaction (after run 1) is given in Fig. 8. Nearly all the perovskite-based catalysts undergo phase transformation to LaCO3OH or mixed La2O(CO3).xH2O and LaCO3OH phases, with only Pt/LaCrO3 remaining structurally stable. The observed breakdown of the LaAlO3 perovskite is in contradiction to previous APR studies utilising the material with supported Ni [26, 27]. This contradiction with the preceding literature is interesting, given the similarity in reaction temperature, the use of a glycerol substrate and the concentration of glycerol being comparable between studies. Differences in stability could be attributable to: (1) the influence of liquid phase product distribution caused by the use of Ni instead of Pt. (2) The residence time of liquid phase products, with the former study being performed in flow and the current being in batch, resulting in longer contact times for the current study. (3) Differences in the synthesis of perovskite, namely a higher calcination temperature was used in prior work (850 °C vs 700 °C) and synthesis method. It is important to note however, that no by-phases were observed in the prepared 700 °C LaAlO3.

XRD patterns of recovered a Pt/Al2O3 (black), Pt/LaAlO3 (red), Pt/LaCrO3 b Pt/LaMnO3 (green), Pt/LaFeO3 (grey), Pt/LaCoO3 (orange), Pt/LaNiO3 (pink) post first reaction. Phases:  γ-Al2O3;
γ-Al2O3;
 LaCO3OH;
LaCO3OH;  perovskite;
perovskite;  La2O(CO3)2·xH2O;
La2O(CO3)2·xH2O;  Fe3O4;
Fe3O4;  Ni
Ni
The XRD pattern of the Pt/LaNiO3 post reaction showed the formation of Ni(0) particles possibly from reduction of Ni from within perovskite structure, phase separated NiO, or residual NiO on reaction surface by in situ generated H2. The reflections for Ni are shifted ((111) shifted from 52.2 to 52.1 º2θ) from literature values potentially indicating alloy formation with Pt. Crystalline M site metal oxide was only observed by XRD in the used LaFeO3 perovskite with Fe3O4 present, alongside LaCO3OH, La2O(CO3)·xH2O, and some residual perovskite phase. No other decomposed perovskites had observable crystalline M site metal or metal oxides present. As previously stated, leaching of Al, Fe, Ni into solution was observed. However, clearly the extent of leaching in Al was not significant enough to account for all the Al and therefore it is anticipated that poorly crystalline γ-Al2O3 or AlO(OH) was formed, which cannot be detected by XRD. Major leaching (> 66%) observed for the Co and Mn catalysts suggests the absence of significant amounts of oxide phases, crystalline or amorphous. The poor conversion observed for the Co and Mn catalysts and lack of their catalytic stability indicates the importance of residual M site oxides in catalytic performance.
Despite the observed phase change, upon further testing, Pt/LaAlO3 and Pt/LaNiO3 maintain stable activity despite structural collapse. No further phase changes are noted in the XRD patterns upon testing up to three cycles (Figure S10). Evidently, these structures decompose into multiple phases in which an active component is present.
XPS analysis of the used catalysts is shown in Table 5 and Figs. 9 and 10. The La environment between fresh and used catalysts show changes in the multiplet splitting from 3.9 to 3.5 eV, which can be assigned to changes from La(OH)3 to La2(CO3)3 [38]. The extensive formation of lanthanum carbonates is consistent with the formation of LaCO3OH and La2O(CO3).xH2O phases seen by XRD. The exception is the La 3d environment in Pt/LaCrO3, which remains unchanged with an assigned to La(OH)3, in agreement with retention of the perovskite structure [37].

XPS spectra of La 3d/Ni 2p and Pt 4f/Al 2p regions for a Pt/LaNiO3, b Pt/LaMnO3, c Pt/LaCrO3, d Pt/LaAlO3, e Pt/Al2O3

XPS Spectrum of Cr 2p region for Pt/LaCrO3
Regarding M site evolution during reaction by XPS the most dramatic change was the absence of any Mn 2p signal. The extent of Mn leaching in the reaction filtrate observed by MP-AES suggests that most Mn is leached from the sample and we hypothesis that any remaining is subsurface. The Cr 2p region post reaction shows reduction of the Cr5+/Cr6+ peak at 654.15 eV with increased intensity of Cr3+ peak at 575.93 eV showing reduction of LaCrO4 by-phase. Further analysis of the Pt/LaCrO3 post 2 and 3 re-use confirm the stability of Pt/LaCrO3 with limited changes in the Cr 2p and La 3d region (Fig. 10). Analysis of the Ni 2p region post reaction show peak at 854.73 eV that can be assigned to Ni2+/Ni3+ species [54]. Interestingly, for the perovskites where La and M concentration could be accurately determined (Cr and Al) a clear inversion of La:M surface concentration was observed post reaction, with surface enrichment of the M site. This is clearly most significant for the structurally stable Pt/LaCrO3 catalyst and indicates that, while the bulk structure is stable, surface reconstruction was observed. Reduction of Pt species during reaction by products, such as H2, is also observed in the Pt 4f region of all the used catalysts, with asymmetric doublets at 70.2–71 eV assigned to Pt(0) [46, 55]. Therefore Pt(0) can be attributed as the active species but no prior reduction step is necessary for its production. Observed Pt(0) species in Pt/LaNiO3 are shifted negatively compared to standards, indicating electronic modification of Pt species by Ni atoms, which has been previously attributed to alloy formation [56]. Except for Pt/LaAlO3 and Pt/γ-Al2O3, surface concentrations of Pt dropped during reaction which suggests aggregation of Pt or surface reconstruction, leading to lower atm % of surface Pt atoms.
XAS of all catalysts after 1st re use found that Pt was mostly reduced to Pt(0) in all catalysts (Table 6). However, linear combination fitting of the XANES and also the required inclusion of Pt–O paths in the EXAFS fits showed that all used catalysts contained a notable oxide component. The exception being for Pt/LaNiO3 which contained limited PtOx (3% by LCF and none noted by XPS). Coordination numbers for 1st shell Pt(0) were found to vary, while in part due to its small contribution to the EXAFS signal, Pt-O coordination numbers were all found be relatively consistent when floated around a value of 1. In several catalysts acceptable fits required fixing of the Pt–O coordination number to values between 1.3 and 1.8. The presence of PtOx in the used catalysts could be attributable to incomplete reduction of Pt under APR reactions due to strong metal support interactions.
Considering the coordination numbers for Pt(0), it is known that for particles < 3 nm values drop below the ideal bulk coordination of 12 in cubic close packed metals such as Pt. It is therefore highly informative that the coordination numbers of ca. 8 were seen for catalysts that showed good catalytic stability (Pt/γ-Al2O3, Pt/LaAlO3/Pt/LaCrO3 and Pt/LaNiO3). While coordination values approach the bulk value of 12 were seen for the catalytically unstable Pt/LaMnO3, Pt/LaFeO3 and Pt/LaCoO3. Evidently, given the oxide component, large Pt(0) particles would not be expected to have a coordination number of 12. Further, particle size distribution cannot be accounted for in EXAFS which is a global averaging technique. However, it does point to the presence of significant atomic fraction of < 3 nm Pt(0) particles in the catalytically stable catalysts. A final and intriguing observation from the EXAFS fitting of the used Pt/LaNiO3 was that fits that used Pt–Pt paths were poor when contrasted to using a Pt-Ni path (Figure S12 and Table S3). EXAFS therefore confirms observations from XRD and XPS analysis that Pt this used catalyst alloys during reaction with Ni exsoluted from the decomposed LaNiO3 support structure.
TEM images taken of the catalysts after several reaction cycles confirm the extent of phase separation of Pt/LaMO-3 into distinct crystals of LaCO3OH and MOx phases after the reaction (TEM of Pt/LaAlO3 used as an exemplar in Fig. 11a). Little preference is noted for Pt nanoparticle migration onto these different phases. Formation of LaCO3OH phases into various morphologies such as triangles or rods are observed within the different catalysts (Figure S13), highlighting the complex mechanisms of perovskite degradation and carbonate phase formation. A single-phase carbonate is observed for Pt/LaMnO3 after two reaction cycles, where the Mn leaches into solution (Figure S13a). Whereas, in the stable Pt/LaCrO3 (Fig. 11b), a similar mixed metal oxide structure as the fresh catalyst is retained (Fig. 4b). Particle size distribution analysis of Pt particles in the used catalysts after three reaction cycles is shown in Fig. 11c–f. Catalysts that undergo phase separations (Pt/LaAlO3 and Pt/LaNiO3) have clear particle growth with large standard deviations, which can be attributed to the uncontrolled breakdown of perovskite structure and complex Pt particle migration mechanisms. Limited particle agglomeration is seen in Pt/Al2O3 and Pt/LaCrO3 where the support materials undergo only surface reconstructions.

TEM images of a Pt/LaAlO3 and b Pt/LaCrO3 and particle size distribution histograms of c Pt/Al2O3, d Pt/LaAlO3, e Pt/LaCrO3, and f Pt/LaNiO3 after three reaction cycles. Particle count of sample size 121–286
From characterisation of post reaction catalysts, it can be hypothesised that the breakdown of the perovskite structure follows M site dissolves into solution or where the M sites undergoes phase separation to the respective single metal oxide (Eqs. 1 and 2). These reactions are potentially mediated by the acidic products and/or through the aggressive hydrothermal conditions of the reaction.
During the APR of glycerol CO2 is produced through the reforming reaction and dissolves in the aqueous conditions to form carbonic acid, which rapidly dissociates to carbonate species [58] (Eq. 3). These carbonate species can undergo a reaction under the hydrothermal conditions with La(OH)3 species to form the LaCO3OH species (Eq. 4) or the La2O(CO3)2·xH2O species (Eq. 5).
It should be noted that the fraction of CO2 sequestered on the formation of lanthanum carbonate phases is very small and has little impact on the observed carbon mass balances. Taking the case for Pt/LaAlO3, the number of moles of La able to sequester CO2 within the reactor is 2.8 × 10−4 mol. Assuming a reaction selectivity of 100% towards the reforming reaction, all lanthanum present will be saturated with carbonate by 0.5% conversion of glycerol.
Reduced Pt species that are clearly responsible for glycerol activation then appear to migrate to the LaCO3OH and MOx support structures, with little preference for either specific support. During this process clear agglomeration of Pt occurred, although perhaps not to the extent expected for the complete collapse of some support structures. It is worth noting the complex mechanisms that must be present to facilitate the migration of these Pt particles, although it is not without precedence, as it is often observed in the restructuring of MgO supported catalysts to Mg(OH)2 in a number of liquid phase biomass transformations [59]. Two clear questions arise from these observations; firstly, how influential are reaction conditions in causing perovskite segregation. Are hydrothermal conditions or the formation of a reducing atmosphere and/or acids products the cause for the observed phase change? Secondly, is the high selectivity towards lactic acid at the expense of reforming caused by the phase change, or because of the subsequently produced Pt/LaCO3OH catalyst?
3.6 Influence of Hydrothermal/Acidic Conditions on Perovskite Supports
To determine stability under the hydrothermal reaction conditions, XRD analysis of the LaMO3 supports (without Pt) after reaction with the glycerol/water solution was performed (Fig. 12). In contrast to the catalysts after the APR reaction, LaAlO3 and LaFeO3 are stable under hydrothermal conditions. Yet, LaMnO3 and LaCoO3 decomposes completely to La(OH)3 and LaCO3OH with crystalline Co(OH)2 and no crystalline Mn phases. LaNiO3 also decomposed completely into LaCO3OH, NiO and Ni, as seen with Pt/LaNiO3. Although the fraction of reduced Ni was far greater for the Pt containing material due to H2 produced in the APR reaction.

XRD patterns of a LaAlO3 (black), LaMnO3 (red); b LaFeO3 (blue), LaCoO3 (green), LaNiO3 (grey) post reaction. Phases:  perovskite;
perovskite;  LaCO3OH;
LaCO3OH; La(OH)3;
La(OH)3;  Co(OH)2;
Co(OH)2; NiO;
NiO;  Ni
Ni
MP-AES analysis of the filtrates of the supports after reaction conditions (Table 7) showed that M site leaching is greatly reduced when compared to the Pt catalysts (Table 4). Despite no crystalline Mn phases, the low amount of Mn in the filtrate suggests the presence of amorphous Mn phases. Interestingly, the limited extent of Co leaching is similar to that in previous studies [53]. However, in contrast to the study by Yang et al., we observe phase separation of LaCoO3 to La(OH)3, LaCO3OH and Co(OH)2.
It is notable that some of the perovskite materials (LaAlO3 and LaFeO3) are stable under hydrothermal conditions, but not as the synthesised Pt/LaMO3 catalysts. The results suggest that for these supported catalysts, reaction products facilitate dissolution of the perovskite structure. To rationalise the observed stability of certain perovskite materials, various factors can be considered. Under reaction conditions, the production of hydrogen in the batch reactor leads to a reducing atmosphere. Therefore, stability at elevated temperatures under reducing conditions must be considered. Nakamura et al. found the order of stability of perovskites under a reducing atmosphere to be: LaCrO3 > LaFeO3 > LaMnO3 > LaNiO3, which can be correlated to the relative M–O bond strengths (Table S4) and initial stability of the M3+cation in the perovskite structure [60]. The stability of LaCrO3 was noted to be due to the crystal-field stabilisation of low spin, t 32g occupied, Cr3 in the octahedral configuration. The stability of LaCrO3 can therefore partly attributed to poor reducibility. Evidently, reducibility is not a sufficient parameter to consider LaAlO3 stability.
Clearly, the stability of the materials under acid conditions must be considered. The pH of post reaction solutions of was measured at room temperature, with Pt supported catalyst solutions having pH values in the range of 2.7–3.3. Much higher pH values of 5.2 and 5.0 observed for the LaCoO3 and LaMnO3 effluents, where Co and Mn leached into solution, alongside a breakdown in the perovskite structure through acid mediated pathways. Van Haasterecht et al. have demonstrated through experimental work and consultation of the relevant Pourbaix diagram, with Ni catalysts, that water and acids are partially oxidising under APR conditions and can facilitate Ni2+ dissolution [61, 62]. According to Pourbaix diagrams (Figures S14–S21), calculated in materials project [63, 64], all the perovskite material studied have limited stability under acid conditions but are stable under alkaline conditions. Al2O3 has some stability under acidic conditions, however, at lower pHs the material is predicted to be unstable (Figure S20), and under APR conditions Al2O3 is known to undergo phase change [16]. Therefore, according to the relevant Pourbaix diagram, the stability of Pt/LaCrO3 catalyst is unexpected alongside the previously reported stability of Ni/LaAlO3 [26, 27]. As observed in studies of oxygen evolution perovskite catalysts within acidic media, potentially the mechanism of dissolution allows for a passivation effect and the formation of stable A or M site enriched surfaces [65]. This corresponds with the formation of M site enriched surfaces by XPS.
To verify the influence of acidic conditions an experiment reported by Van Haasterecht et al., where the addition of base (KOH) improved an Ni/C catalysts activity and stability, was emulated [62]. 0.5 M KOH was added to the glycerol stock solution (pH 13.1) and investigated for APR using the Pt/LaAlO3 catalyst. As observed by Van Haasterecht et al., activity increased, with reduction in CO2 selectivity, possibly due to it being sequestered by KOH, and an increased hydrogen selectivity (Table 8). Substantial enhancement in lactic acid production was observed upon base addition (Fig. 13).

Carbon product selectivity comparison for APR of glycerol over Pt/LaAlO3 with or without added KOH. Products: Lactic Acid (LA), Ethylene Glycol (EG), Hydroxyacetone (HA), 1,2-Propanediol, 2-Propanol (2-P), 1-Propanol (1-P), and Ethanol (E). Reaction conditions: 240 °C, PAr = 42 bar, 1000 RPM, 20 ml 10 wt% glycerol, 0.5 M KOH, 60 mg catalyst, 2 h
The pH of the effluent post reaction increased from 3.0, observed in the absence of base, to 7.6. Characterisation of the recovered catalyst by XRD (Fig. 14) showed a substantial increase in perovskite structure stability, although clear reflections with La2O(CO3)2·xH2O were also observed. As predicted by the Pourbaix diagram perovskite stability could be significantly enhanced by maintaining alkaline conditions. Potentially using flow reactors, pH control and contact time could be controlled to a sufficient extent to produce structurally stable catalysts.

XRD pattern of recovered Pt/LaAlO3 post reaction with added 0.5 M KOH. Phases:  perovskite;
perovskite;  La2O(CO3)2·xH2O
La2O(CO3)2·xH2O
3.7 Pt/LaCO3OH
Finally, the potential of Pt supported on LaCO3OH, the by-product of LaMO3 decomposition was briefly considered. Previous studies of liquid phase hydrogenolysis of glycerol using Ru/LaCO3OH catalysts show high stability and activity of LaCO3OH materials [32]. LaCO3OH with a surface area of 13 m2g−1 was synthesised and Pt successfully deposited. The catalyst showed good conversion (15%), low conversion to gas (7%) and high lactic acid selectivity (31%). Carbon mass balance was 97%. The high lactic acid selectivity observed was similar to the Pt/LaMnO3 and Pt/LaCoO3 catalysts (39 and 38% respectively) which leach the M site metal during reaction. Therefore, it can be concluded that Pt/LaCO3OH shows particularly high selectivity towards lactic acid relative to the standard Pt/γ-Al2O3. The higher conversion observed relative to perovskite supported catalysts, in which M site leaching was observed, can be attributed to the uncontrolled redistribution of Pt during reaction in the latter case. The potential of these LaCO3OH catalysts for the production of hydrogen through dehydrogenation and reactions with limited CO2 production requires dedicated further study.
4 Conclusions
The stability and activity of Pt/LaMO-3 catalysts (where M = Al, Cr, Mn, Fe, Co, Ni) have been evaluated for the aqueous phase reforming of glycerol at 240 °C, 42 bar. Most of the perovskite catalysts break down into LaCO3OH and the respective M oxides, or in the case of Mn and Co, forming water soluble complexes and leaching into the reaction. An exception was the Pt/LaCrO3 catalyst, which was stable under reaction conditions, with improved activity and hydrogen productivity when compared to a standard Pt/γ-Al2O3 catalyst. While all other supports decomposed, catalytic stability was observed for Pt/LaAlO3 and Pt/LaNiO3, due to the redistribution of Pt over the formed LaCO3OH and the respective M oxides. In the case of Pt/LaNiO3 there was evidence of PtNi alloy formation under reaction conditions. All LaMO3 supported catalysts showed high selectivity towards lactic acid, which was attributed to the Lewis basic LaCO3OH or LaCrO3 phases. However, a lack of control over Pt dispersion, as seen by large particle size standard deviations, over various phases on perovskite decomposition make most of these catalysts practically undesirable. The exception being Pt/LaCrO3. Stability can be correlated to various factors, which need to be considered when designing catalysts for APR. These include acid stability, which has been preliminarily investigated using Pourbaix diagrams, stability of materials under reducing conditions, and M–O bond strength. Perovskite support stability could be improved by the addition of KOH base to the reaction, which also dramatically increased reaction rates and hydrogen production. However, while significant amount of perovskite was observed by XRD post reaction, other phases were observed demonstrating decomposition rates had been reduced as opposed to being stopped. Intentionally prepared Pt/LaCO3OH catalysts were found to be active and stable in the APR reaction and the stability of materials warrants further study.
References
Baloat M (2008) Potential importance of hydrogen as a future solution to environmental and transportation problems. Int J Hydrogen Energy 33:4013–4029
Mazloomi K, Gomes C (2012) Hydrogen as an energy carrier: prospects and challenges. Renew Sustain Energy Rev 16:3024–3033
Abe JO, Popoola API, Ajenifuja E, Popoola OM (2019) Hydrogen energy, economy and storage: review and recommendation. Int J Hydrogen Energy 44:15072–15086
Muradov N, Vezirolu T (2005) From hydrocarbon to hydrogen-carbon to hydrogen economy. Int J Hydrogen Energy 30:225–237
Davda RR, Shabaker JW, Huber GW, Cortright RD, Dumesic JA (2003) Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts. Appl Catal B 43:13–26
Shabaker JW, Huber GW, Davda RR, Cortright RD, Dumesic JA (2003) Aqueous-phase reforming of ethylene glycol over supported platinum catalysts. Catal Lett 88:1–8
Coronado I, Stekrova M, Reinikainen M, Simell P, Lefferts L, Lehtonen J (2016) A review of catalytic aqueous-phase reforming of oxygenated hydrocarbons derived from biorefinery water fractions. Int J Hydrogen Energy 41:11003–11032
Seretis A, Tsiakaras P (2015) A thermodynamic analysis of hydrogen production via aqueous phase reforming of glycerol. Fuel Process Technol 134:107–115
Ripken RM, Meuldijk J, Gardeniers JGE, Le Gac S (2017) Influence of the water phase state on the thermodynamics of aqueous-phase reforming for hydrogen production. Chemsuschem 10:4909–4913
Huber GW, Shabaker JW, Dumesic JA (2003) Raney Ni-Sn catalyst for H2 production from biomass-derived hydrocarbons. Science 300:2075–2077
Gogoi P, Nagpure AS, Kandasamy P, Satyanarayana CVV, Raja T (2020) Insights into the catalytic activity of Ru/NaY catalysts for efficient H2 production through aqueous phase reforming. Sustain Energy Fuels 4:678–690
Liu J, Sun B, Hu J, Pei Y, Li H, Qiao M (2010) Aqueous-phase reforming of ethylene glycol to hydrogen on Pd/Fe 3O4 catalyst prepared by co-precipitation: metal-support interaction and excellent intrinsic activity. J Catal 274:287–295
Davda RR, Shabaker JW, Huber GW, Cortright RD, Dumesic JA (2005) A review of catalytic issues and process conditions for renewable hydrogen and alkanes by aqueous-phase reforming of oxygenated hydrocarbons over supported metal catalysts. Appl Catal B 56:171–186
Subramanian ND, Callison J, Catlow CRA, Wells PP, Dimitratos N (2016) Optimised hydrogen production by aqueous phase reforming of glycerol on Pt/Al2O3. Int J Hydrogen Energy 41:18441–18450
Luo N, Fu X, Cao F, Xiao T, Edwards PP (2008) Glycerol aqueous phase reforming for hydrogen generation over Pt catalyst—effect of catalyst composition and reaction conditions. Fuel 87:3483–3489
Koichumanova K, Sai Sankar Gupta KB, Lefferts L, Mojet BL, Seshan K (2015) An in situ ATR-IR spectroscopy study of aluminas under aqueous phase reforming conditions. Phys Chem Chem Phys 17:23795–23804
Liu F, Okolie C, Ravenelle RM, Crittenden JC, Sievers C, Bruijnincx PCA, Weckhuysen BM (2018) Silica deposition as an approach for improving the hydrothermal stability of an alumina support during glycerol aqueous phase reforming. Appl Catal A 551:13–22
Lei Y, Lee S, Bin Low K, Marshall CL, Elam JW (2016) Combining electronic and geometric effects of ZnO-promoted Pt nanocatalysts for aqueous phase reforming of 1-propanol. ACS Catal 6:3457–3460
Guo Y, Azmat MU, Liu X, Wang Y, Lu G (2012) Effect of support’s basic properties on hydrogen production in aqueous-phase reforming of glycerol and correlation between WGS and APR. Appl Energy 92:218–223
Chen G, Xu N, Li X, Liu Q, Yang H, Li W (2015) Hydrogen production by aqueous-phase reforming of ethylene glycol over a Ni/Zn/Al derived hydrotalcite catalyst. RSC Adv 5:60128–60134
Li D, Li Y, Liu X, Guo Y, Pao C-W, Chen J-L, Hu Y, Wang Y (2019) NiAl2O4 spinel supported pt catalyst: high performance and origin in aqueous-phase reforming of methanol. ACS Catal 9:9671–9682
Morales-Marín A, Ayastuy JL, Iriarte-Velasco U, Gutiérrez-Ortiz MA (2019) Nickel aluminate spinel-derived catalysts for the aqueous phase reforming of glycerol: effect of reduction temperature. Appl Catal B 244:931–945
Reynoso AJ, Ayastuy JL, Iriarte-Velasco U, Gutiérrez-Ortiz MA (2018) Cobalt aluminate spinel-derived catalysts for glycerol aqueous phase reforming. Appl Catal B 239:86–101
Tejuca LG, Fierro G, Tacon JMD (1989) Structure and reactivity of perovskite-type oxides. Adv Catal 36:237–328
Royer S, Duprez D, Can F, Courtois X, Batiot-Dupeyrat C, Laassiri S, Alamdari H (2014) Perovskites as substitutes of noble metals for heterogeneous catalysis: dream or reality. Chem Rev 114:10292–10368
Park YH, Kim JY, Moon DJ, Park NC, Kim YC (2015) Effect of LaAlO3-supported modified Ni-based catalysts on aqueous phase reforming of glycerol. Res Chem Intermed 41:9603–9614
Lee H-J, Shin GS, Kim Y-C (2015) Characterization of supported Ni catalysts for aqueous-phase reforming of glycerol. Korean J Chem Eng 32:1267–1272
Zhu H, Zhang P, Dai S (2015) Recent advances of lanthanum-based perovskite oxides for catalysis. ACS Catal 5:6370–6385
Evans CD, Kondrat SA, Smith PJ, Manning TD, Miedziak PJ, Brett GL, Armstrong RD, Bartley JK, Taylor SH, Rosseinsky MJ, Hutchings GJ (2016) The preparation of large surface area lanthanum based perovskite supports for AuPt nanoparticles: tuning the glycerol oxidation reaction pathway by switching the perovskite B site. Faraday Discuss 188:427–450
Hwang J, Rao RR, Giordano L, Katayama Y, Yu Y, Shao-Horn Y (2017) Perovskites in catalysis and electrocatalysis. Science 358:751–756
Deganello F, Marcì G, Deganello G (2009) Citrate–nitrate auto-combustion synthesis of perovskite-type nanopowders: a systematic approach. J Eur Ceram Soc 29:439–450
Li B, Li L, Zhao C (2017) A highly stable Ru/LaCO 3 OH catalyst consisting of support-coated Ru nanoparticles in aqueous-phase hydrogenolysis reactions. Green Chem 19:5412–5421
Newville M (2001) IFEFFIT: interactive XAFS analysis and FEFF fitting. J Synchrotron Radiat 8:322–324
Ravel B, Newville M (2005) ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Radiat 12:537–541
Carter JD, Anderson HU, Shumsky MG (1996) Structure and phase transformation of lanthanum chromate. J Mater Sci 31:551–557
Lima SM, Assaf JM, Peña MA, Fierro JLG (2006) Structural features of La1-xCexNiO3 mixed oxides and performance for the dry reforming of methane. Appl Catal A 311:94–104
Uwamino Y, Ishizuka T, Yamatera H (1984) X-ray photoelectron spectroscopy of rare-earth compounds. J Electron Spectros Relat Phenomena 34:67–78
Li JPH, Zhou X, Pang Y, Zhu L, Vovk EI, Cong L, van Bavel AP, Li S, Yang Y (2019) Understanding of binding energy calibration in XPS of lanthanum oxide by in situ treatment. Phys Chem Chem Phys 21:22351–22358
Biesinger MC, Payne BP, Grosvenor AP, Lau LWM, Gerson AR, Smart RSC (2011) Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl Surf Sci 257:2717–2730
Ponce S, Peña MA, Fierro JLG (2000) Surface properties and catalytic performance in methane combustion of SR-substituted lanthanum manganites. Appl Catal B 24:193–205
Parida KM, Reddy KH, Martha S, Das DP, Biswal N (2010) Fabrication of nanocrystalline LaFeO3: an efficient sol-gel auto-combustion assisted visible light responsive photocatalyst for water decomposition. Int J Hydrogen Energy 35:12161–12168
Munakata F, Takahashi H, Akimune Y, Shichi Y, Tanimura M, Inoue Y (1997) Electronic state and valence control of LaCoO3: difference between La-deficient and Sr-substituting effects. Phys Rev B 56:979–982
Rida K, Benabbas A, Bouremmad F, Peña MA, Sastre E, Martínez-Arias A (2007) Effect of calcination temperature on the structural characteristics and catalytic activity for propene combustion of sol–gel derived lanthanum chromite perovskite. Appl Catal A 327:173–179
Pereñíguez R, González-DelaCruz VM, Holgado JP, Caballero A (2010) Synthesis and characterization of a LaNiO3 perovskite as precursor for methane reforming reactions catalysts. Appl Catal B 93:346–353
Stojanović M, Haverkamp R, Mims C, Moudallal H, Jacobson A (1997) Synthesis and characterization of LaCr1 − xNixO3 perovskite oxide catalysts. J Catal 166:315–323
Bancroft GM, Adams I, Coatsworth LL, Bennewitz CD, Brown JD, Westwood WD (1975) ESCA study of sputtered platinum films. Anal Chem 47:586–588
Wawrzetz A, Peng B, Hrabar A, Jentys A, Lemonidou AA, Lercher JA (2010) Towards understanding the bifunctional hydrodeoxygenation and aqueous phase reforming of glycerol. J Catal 269:411–420
Callison J, Subramanian ND, Rogers SM, Chutia A, Gianolio D, Catlow CRA, Wells PP, Dimitratos N (2018) Directed aqueous-phase reforming of glycerol through tailored platinum nanoparticles. Appl Catal B 238:618–628
Kishida H, Jin F, Zhou Z, Moriya T, Enomoto H (2005) Conversion of glycerin into lactic acid by alkaline hydrothermal reaction. Chem Lett 34:1560–1561
Chen L, Ren S, Ye XP (2014) Lactic acid production from glycerol using CaO as solid base catalyst. Fuel Process Technol 120:40–47
Polo-Garzon F, Wu Z (2018) Acid–base catalysis over perovskites: a review. J Mater Chem A 6:2877–2894
Shabaker JW, Simonetti DA, Cortright RD, Dumesic JA (2005) Sn-modified Ni catalysts for aqueous-phase reforming: characterization and deactivation studies. J Catal 231:67–76
Yang X, Yang L, Fan W, Lin H (2016) Effect of redox properties of LaCoO3 perovskite catalyst on production of lactic acid from cellulosic biomass. Catal Today 269:56–64
Grosvenor AP, Biesinger MC, Smart RSC, McIntyre NS (2006) New interpretations of XPS spectra of nickel metal and oxides. Surf Sci 600:1771–1779
Kim KS, Winograd N, Davis RE (1971) Electron spectroscopy of platinum-oxygen surfaces and application to electrochemical studies. J Am Chem Soc 93:6296–6297
Kong F, Ren Z, Norouzi Banis M, Du L, Zhou X, Chen G, Zhang L, Li J, Wang S, Li M, Doyle-Davis K, Ma Y, Li R, Young A, Yang L, Markiewicz M, Tong Y, Yin G, Du C, Luo J, Sun X (2020) Active and stable Pt-Ni alloy octahedra catalyst for oxygen reduction via near-surface atomical engineering. ACS Catal 10:4205–4214
Shu Y, Murillo LE, Bosco JP, Huang W, Frenkel AI, Chen JG (2008) The effect of impregnation sequence on the hydrogenation activity and selectivity of supported Pt/Ni bimetallic catalysts. Appl Catal A 339:169–179
Adamczyk K, Premont-Schwarz M, Pines D, Pines E, Nibbering ETJ (2009) Real-time observation of carbonic acid formation in aqueous solution. Science 326:1690–1694
Brett GL, He Q, Hammond C, Miedziak PJ, Dimitratos N, Sankar M, Herzing AA, Conte M, Lopez-Sanchez JA, Kiely CJ, Knight DW, Taylor SH, Hutchings GJ (2011) Selective oxidation of glycerol by highly active bimetallic catalysts at ambient temperature under base-free conditions. Angew Chemie 123:10318–10321
Nakamura T, Petzow G, Gauckler LJ (1979) Stability of the perovskite phase LaBO3 (B = V, Cr, Mn, Fe, Co, Ni) in reducing atmosphere I. Experimental results. Mater Res Bull 14:649–659
Patel AM, Nørskov JK, Persson KA, Montoya JH (2019) Efficient Pourbaix diagrams of many-element compounds. Phys Chem Chem Phys 21:25323
Van Haasterecht T, Ludding CCI, De Jong KP, Bitter JH (2014) Toward stable nickel catalysts for aqueous phase reforming of biomass-derived feedstock under reducing and alkaline conditions. J Catal 319:27–35
Persson KA, Waldwick B, Lazic P, Ceder G (2012) Prediction of solid-aqueous equilibria: scheme to combine first-principles calculations of solids with experimental aqueous states. Phys Rev B 85:
Jain A, Ong SP, Hautier G, Chen W, Richards WD, Dacek S, Cholia S, Gunter D, Skinner D, Ceder G, Persson KA (2013) Commentary: the Materials Project: A materials genome approach to accelerating materials innovation. APL Mater 1:
Raman AS, Patel R, Vojvodic A (2020) Surface stability of perovskite oxides under OER operating conditions: A first principles approach. Faraday Discuss
Acknowledgements
We would like to acknowledge funding form the EPSRC CDT “fuel cells and their fuels” (EP/L015749/1). UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grant: EP/R026939/1, EP/R026815/1, EP/R026645/1, EP/R027129/1. The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195. We would like to thank the Cardiff University electron microscopy facility for the transmission (TEM) and scanning electron microscopy (SEM). XAFS measurements were performed at Diamond Light Source on beamline B18 through the UK Catalysis BAG (SP198590-11). We thank Dr. Diego Gianolio and Dr Martin Wilding for their support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Inns, D.R., Mayer, A.J., Skukauskas, V. et al. Evaluating the Activity and Stability of Perovskite LaMO3-Based Pt Catalysts in the Aqueous Phase Reforming of Glycerol. Top Catal 64, 992–1009 (2021). https://doi.org/10.1007/s11244-021-01449-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-021-01449-6