
Abstract
Cu/ZnO methanol catalysts were deposited over several ZSM-5 acid zeolites to directly synthesise oxygenates (methanol and dimethyl ether) from a CO2/H2 feed. Catalysts were prepared by two different preparation methodologies: chemical vapour impregnation (CZZ-CVI) and oxalate gel precipitation (CZZ-OG). Chemical vapour impregnation led to Cu/ZnO being deposited on the zeolite surface, whilst oxalate gel precipitation led to the formation of Cu/ZnO agglomerates. For both sets of catalysts a higher concentration of mild and strong acid sites were produced, compared to the parent ZSM-5 zeolites, and CZZ-CVI had a higher concentration of acid sites compared to CZZ-OG. Nevertheless, CZZ-OG shows considerably higher oxygenate productivity, 1322 mmol Kgcat−1 h−1, compared to 192 mmol Kgcat−1 h−1 over CZZ-CVI (ZSM-5(50), 250 ℃, 20 bar, CO2/H2 = 1/3, 30 ml min−1), which could be assigned to a combination of smaller particle size and enhanced methanol mass transfer within the zeolites.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
CO2 hydrogenation allows us to store hydrogen produced from renewable resources (green hydrogen) in the form of liquid fuels [1]. Thus, green hydrogen [2] can be readily incorporated into the current energy infrastructure, while prominent technologies (e.g. PEM electrolysers) which use hydrogen as an energy vector are well-established. Among the possible CO2 hydrogenation products [3], CH3OH is of great interest [4]. CH3OH can be blended with gasoline or converted into gasoline. For instance, in 1985 to meet the domestic fuel demand in New Zealand two methanol plants were combined with the methanol-to-gasoline process developed by Mobil [5]. Moreover, CH3OH is also used as an intermediate for the production of numerous important chemicals (e.g. ethylene, propylene) [6]. CH3OH is currently produced over Cu/ZnO/Al2O3 catalysts from syngas (CO + H2 + CO2) at 200–300 °C and 80–120 bar [5]. When CO2 (Eq. 1) is the feed instead of CO (Eq. 2) the CH3OH yield is constrained by equilibrium. For instance, at 250 °C and 30 bar, CH3OH yield is limited to ca. 12 C-mol %, while CH3OH yield increase to ca. 42 C-mol % using a CO/CO2 = 4 feed (H2/(CO + CO2) = 3) [7]. Under laboratory testing conditions (20–50 bar) the CO2 hydrogenation to CH3OH is limited to the 200–270 °C range, at lower temperature little CO2 conversion is observed, while at higher CH3OH productivity is limited by equilibrium whilst the reverse water gas shift (RWGS) reaction is promoted (Eq. 3) [8].
CH3OH productivity from CO2 can be enhanced by simultaneously dehydrating CH3OH to dimethyl ether (DME) (Eq. 4) by combining a CH3OH synthesis catalyst with a solid acid catalyst (overall reaction in Eq. 5), known as hybrid catalysts. Like CH3OH, DME synthesis from CO2 is favoured at low temperature and high pressure. DME synthesis from CO2 is advantageous compared to CH3OH because of the higher DME equilibrium selectivity. Moreover, DME can be employed as a fuel in diesel engines because of its higher cetane number and lower auto-ignition temperature compared to diesel fuel (55–60 compared to 40–50, and 235 °C compared to 250 °C, respectively), and can be easily liquified at low pressure, hence it can be used as a liquified petroleum gas (LPG) substitute [9, 10].
Based on the actual CH3OH synthesis process (developed by Imperial Chemical Industries), most of the research on the CO2 hydrogenation to CH3OH focus on Cu/ZnO catalysts [11]. As well summarised by Spencer [12], ZnO prevents the sintering of Cu particles by acting as a refractory spatial binder, maintaining small Cu crystallites from agglomerating during reaction [13]. Nevertheless, ZnO not only plays a spatial role in improving the catalyst lifespan, but it is also an activity promoter. As Spencer elucidated: on one hand, under CH3OH synthesis conditions surface CuZn alloys can form, leading to the formation of Cu+–O–Zn active sites [14,15,16]. On the other hand, under laboratory testing conditions (moderate pressure, CO2/H2 = 1/3, high conversion) H2 dissociates on ZnO and spills over onto Cu particles [17]. For the CH3OH dehydration to dimethyl ether (DME) ZSM-5 zeolites are preferred to ɣ-alumina as the acid catalyst because ZSM-5 zeolites contain Lewis and Brønsted acid sites, their high resistance to water, and their chemical and thermal stability [11]. For instance, Aguayo et al. [18] compared Cu–ZnO–Al2O3/ɣ-Al2O3 and Cu–ZnO–Al2O3/NaZSM-5 hybrid catalysts for CH3OH synthesis from the hydrogenation of CO and CO2. It was observed that competitive adsorption between H2O and CH3OH for acid sites on ɣ-Al2O3, leading to lower CO/CO2 conversion and DME selectivity compared to ZSM-5. Moreover, catalyst deactivation after regeneration was observed for the ɣ-Al2O3-containing hybrid catalyst, whilst good regeneration was observed for the Cu–ZnO–Al2O3/ZSM-5 hybrid catalyst. Similar conclusions were reported by Naik et al. [19] Bonura and co-workers studied the effect of water on CH3OH dehydration activity over ZSM-5 zeolites. At low temperature H2O competes with acid sites resulting in a decrease in CH3OH conversion, with higher deactivation observed with increasing the zeolite acidity. Whilst, no appreciable decrease in CH3OH dehydration was observed above 180 °C [20]. Readers are referred to reports by Catizzone et al. [21] and Álvarez et al. [11] for a detailed review on the CO2 upgrading to DME.
Frusteri et al. [20] compared the activity of CuZnZr/ZSM-5 hybrid catalysts with CuZnZr supported over ZSM-5 by co-precipitation. Catalysts where the CH3OH synthesis catalyst is supported over the solid acid catalyst are known as integrated catalysts. Slightly higher CO2 conversion and DME productivity was observed over the integrated catalyst, which was attributed to the spatial proximity between the two active functionalities. Due to the benefits of integrated catalysts over hybrid catalysts, the aim of this work is to compare DME productivity between CuZnO/ZSM-5 integrated catalysts prepared via chemical vapour impregnation (CVI) or via oxalate gel precipitation (OG).
2 Experimental Section
2.1 Materials
All materials employed in this work were purchased from chemical suppliers and used as received without prior purification. ZSM-5 zeolites in the (NH4+) form were purchased from Alfa Aesar (Si/Al = 23, 50, 80); Zn acetylacetonate (Zn(acac)2, 99%), Cu acetylacetonate (Cu(acac)2, 97%), Cu nitrate hydrate (Cu(NO3)2·3H2O, 99%) and Zn nitrate hydrate (Zn(NO3)2·6H2O, 99%) and ethanol (99.8%) were purchased from Sigma-Aldrich.
2.2 Catalysts Synthesis
CuZnO/ZSM-5 catalysts synthesised via Chemical Vapour Impregnation (CZZ-CVI). For the preparation of 3.0 g of CuZnO/ZSM-5 catalyst by CVI (20 wt.%, Cu/Zn mol = 1) 1.24 g of Cu(acac)2, 1.21 g Zn(acac)2 and 2.4 g of activated H-ZSM-5 (static air, 550 ℃, 5 ℃ min−1, 4 h) were thoroughly mixed in a glass vial. The homogeneous mixture was then transferred into a Schlenk flask, evacuated (10–3 mbar) and heated to 120 ℃ for 1 h. Samples were then cooled to 30 ℃, recovered and annealed in static air (500 ℃, 2 °C min−1, 16 h).
CuZnO/ZSM-5 catalysts synthesised via Oxalate Gel precipitation (CZZ-OG). The procedure for the preparation of 3.0 g of CuZnO/ZSM-5 (20 wt.%, Cu/Zn mol = 1) catalysts by OG is as follows; 1.14 g of Cu(NO3)2 and 1.37 g of Zn(NO3)2 were dissolved in 200 ml of ethanol, under vigorous stirring 2.4 g of activated H-ZSM-5 (static air, 550 ℃, 5 ℃ min−1, 4 h) were added. 2.16 g of oxalic acid were added to the slurry. The precipitate was aged for 1 h under stirring, recovered by filtration, dried (static air, 100 ℃, 16 h) and annealed (static air, 500 ℃, 10 ℃ min−1, 2 h).
2.3 Catalyst Characterisation
Prior to characterisation a portion of the CZZ catalysts were reduced under flowing 5% H2/Ar (220 ℃, 2 ℃ min−1, 1 h). Powder X-ray diffraction (XRD) patterns were recorded on a (θ-θ) PANalyticalX’pert Pro powder diffractometer fitted with a position sensitive detector using a Cu Kα radiation source (40 keV, 40 mA). Transmission electron microscopy (TEM) images were obtained on a JEOL 2100 (LaB6) instrument fitted with a Gatan Ultrascan 1000xp digital camera operated at 200 kV. Specimens were dry-prepared on 300 mesh copper TEM-grids coated with holey carbon film. Pore size and apparent BET surface area were obtained from N2 adsorption isotherms at (− 196 ℃) on a Quantachrome Nova 2200e instrument. Prior to analysis samples were degassed in situ (120 ℃, 10 h). Ammonia temperature programmed desorption (NH3-TPD) were acquired on a Quantachrome ChemBET TPR/TPD equipped with a TCD detector. 150 mg of pre-reduced catalyst were secured with quartz wool. Prior to analysis samples were heated under a He flow (130–140 ml min−1) at 400 ℃ (10 ℃ min−1, 1 h). The sample cell was cooled to 30 ℃, the flow was then switched to 10% NH3/Ar. After 15 min the gas was switched back to He, heated to 100 ℃ (10 ℃ min−1, 1 h) to remove physiosorbed NH3, and cooled to 50 °C. NH3-TPD profiles were then measured (50–700 ℃, 10 ℃ min−1).
2.4 Catalyst Performance for Oxygenates Synthesis from CO2 and H2
The CO2 hydrogenation and consecutive CH3OH dehydration to DME was assessed in a stainless steel fixed-bed continuous flow reactor (50 cm length, 0.5 cm internal diameter). Temperature was controlled through a chromel–alumel thermocouple placed inside the catalyst bed, pressure was controlled using a back pressure regulator. Catalysts were pressed (10 ton, 1 min) into a 1.3 cm die, the die was then crushed, and sieved into (425–600 µm) pellets. 0.5 g of catalyst pellets were placed in the reactor tube without diluent, quartz wool was used to secure the catalyst bed in place. Prior to reaction catalysts were pre-reduced in situ with 5% H2/Ar (1 bar, 30 ml min−1, 220 °C, 2 °C min−1, 1 h). After pre-reduction, the reactor was cooled down to 50 °C, the gas flow was switched to the reaction mixture (CO2/H2/N2 = 1/3/1), the reactor was pressurised to 20 bar and heated to the desired reaction temperature (230–310 °C). Post-reactor lines and valves were heated at 130 °C to avoid product condensation. Catalytic activity was simultaneously compared against a blank reactor tube under the same reaction conditions. Products were analysed via online GC analysis (Agilent 7890, fitted with FID and TCD detectors).
3 Results and Discussion
3.1 Catalyst Characterisation
CuZnO/ZSM-5 integrated catalysts (20 wt.% Cu and Zn, Cu/Zn mol = 1) were prepared via oxalate gel precipitation (OG) and chemical vapour impregnation (CVI) over commercially sourced ZSM-5 zeolites (Si/Al = 80, 50 and 23). Precipitation of Cu and Zn nitrate precursors in the presence of carbonates or hydroxides is commonly reported for the preparation of CH3OH synthesis catalysts [11], however, physicochemical properties strongly depend on preparation parameters such as pH, temperature, precipitation rate, ageing and drying [22,23,24]. Instead, the oxalate gel precipitation method is reported to yield small, well dispersed nanoparticles with high Cu surface area and homogeneous Cu/Zn distribution [25, 26]. This method was employed to prepare CuZnO/ZSM-5 integrated catalysts (CZZ-OG). CVI was also used for the preparation of CuZnO/ZSM-5 integrated catalysts (CZZ-CVI), because it allows the preparation of well dispersed bimetallic catalysts, and can be used over a wide range of supports, avoids the use of chloride metal precursors and avoids the presence of possible impurities from the use of solvents, ligands and reducing/precipitating agents [27,28,29,30]. Catalyst characterisation was performed on a portion of the pre-reduced sample (5% H2/Ar, 220 °C, 2 °C min−1, 1 h).
The porous structure of commercial ZSM-5 (Si/Al = 23, 50 and 80) zeolites after activation (static air, 550 °C, 5 °C min−1, 4 h) was determined by N2 adsorption. Due to the limitations of using N2 as adsorbent for the analysis of porous materials (microporous filling due to strong adsorption at low p/p0, non-uniform monolayer structure leading to different molecular N2 areas) [31] and that commercially supplied zeolites were employed, values reported in this work were used to qualitatively compare materials. Comparable pore size (ca. 5.5 Å) and apparent BET surface area (ca. 460 m2 g−1) were measured for all three zeolites (Table S1). A slight decrease in the pore size was observed for CZZ-OG and CZZ-CVI catalysts, nevertheless, the BET surface area of the integrated catalysts was significantly lower than the parent zeolites (Table 1). Suggesting that Cu/ZnO nanoparticles were mainly at the surface and not within the zeolite pore structure. BET values are representative of the average surface area of the catalyst, for CZZ catalysts reported in this work, ZSM-5 zeolites were loaded with a ca. 9.7 wt.% Cu and ca. 10.3 wt.% Zn. Hence, a decrease in the surface area is expected, and not indicative of the metal distribution within the pore structure.
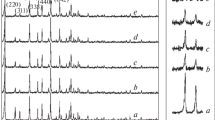
X-ray powder diffraction (XRD) was employed to determine the crystalline phases present in integrated catalysts after pre-reduction. For comparison, XRD patterns for CZZ(23)-OG, CZZ(23)-CVI and ZSM-5(23) are shown in Fig. 1 (XRD patterns for all integrated catalysts and activated ZSM-5 zeolites can be found in Figure S1 and Figure S2, respectively). The presence of metallic Cu (PDF 01-070-3038) was observed at 43.2°, 50.3° and 73.9°. Reflections not assigned to ZSM-5(23) (MFI morphology PDF 00-89-1421) or to Cu were attributed to the presence of ZnO (PDF 00-036-1451). Sharper Cu and ZnO peaks were observed for integrated catalysts prepared by CVI, compared to catalysts prepared by OG, indicating that the former methodology yielded larger crystallites. Oxalate gel co-precipitation has been reported to yield small and well distributed Cu/ZnO [25, 26], however, differences in the crystallite size between CVI and OG could be also attributed to the difference in annealing treatment. The former was annealed at 500 °C for 16 h to ensure the complete decomposition of the acetylacetonate precursors, whilst for the latter the annealing at 500 °C was limited to 2 h to avoid unnecessary crystallite growth. No Cu2O (PDF 01-071-3645) or CuO (PDF 00-048-1548) peaks at 36.4° or 35.6° were observed. The Cu and ZnO crystallite size for CZZ-OG and CZZ-CVI catalysts was estimated using the Scherrer equation (Table 2). Details on how the Scherrer equation was applied to measure Cu and ZnO crystallite sizes can be found in the supporting information (Equation S1–S2). In accordance with N2 adsorption measurements, no significant changes in the pore size were observed between parent zeolites and integrated catalysts, and the measured Cu and ZnO crystallite sizes were considerably larger than the zeolite pores. Hence Cu and ZnO crystallites are physically unable to fit inside the zeolite pores. It was also observed that the oxalate gel precipitation yielded five-fold smaller Cu crystallites compared to CVI.

XRD patterns for H-ZSM-5(23), CZZ(23)-OG and CZZ(23)-CVI after reduction (5% H2/Ar, 220 °C, 2 °C min−1, 1 h)
Morphological differences between CZZ(23)-OG and CZZ(23)-CVI integrated catalysts were analysed by TEM. Figure S3a and Figure S3b show low magnification TEM images for CZZ(23)-OG, it can be observed that most of the Cu and ZnO is not supported at the surface of the ZSM-5(23) zeolite, but instead forms large phase separated aggregates (nm–µm). These large aggregates contradict the Cu crystallite size determined by XRD through the Scherrer equation. However, as shown in Fig. 2a, the large aggregates are made of smaller nanoparticles, in the size range determined by XRD (ca. 15 nm as shown in Fig. 2b).

TEM imaged for CZZ(23)-OG showing (a) large aggregates of Cu and ZnO made of smaller nanoparticles and (b) size of the Cu and ZnO nanoparticles
For CZZ(23)-CVI catalyst two distinct cases are observed. The Cu/ZnO nanoparticles are not well dispersed over every zeolite particle. Zeolite particles either display a complete absence of Cu/ZnO particles (Fig. 3a) or are covered with small and well-dispersed Cu/ZnO nanoparticles (Fig. 3b). Prolonged exposure of the “particle free” zeolite areas under the electron beam led to the formation of Cu/ZnO aggregates on the zeolite surface (Figure S4a to S4c). This behaviour was not observed for the CZZ-OG sample, furthermore, the zeolite showed no morphological change under the same beam conditions. This suggests that the Cu/ZnO is deposited as a film over the surface or as sub-nm particles or clusters inside the zeolite porous framework which are beyond the resolution of the microscope used, and were formed by beam induced sintering, which could be related to the large Cu and ZnO crystallite sizes observed by XRD analysis.

TEM images for CZZ(23)-CVI showing (a) large areas of support which appears to be metal free and (b) small and evenly distributed Cu and ZnO nanoparticles
The CH3OH dehydration to DME is an acid catalysed reaction. Because of the presence of Brønsted and Lewis acid sites zeolites are less prone to deactivation than Al2O3 during CH3OH dehydration. This is explained by the higher affinity of Brønsted acid sites towards CH3OH compared to water [32]. The acidity of zeolites is an important parameter to optimise CH3OH dehydration. Increasing the Al concentration within the structure strengthens the acidity, this leads to improved CH3OH conversion, but usually at the expense of DME selectivity towards the production of other hydrocarbons, and decreases catalyst lifetime due to coke deposition [33]. NH3 is normally employed to characterise the acid properties of porous materials because of its strong basicity and small molecular size. Ammonia temperature programmed desorption (NH3-TPD) was employed to compare acid properties of activated zeolites and Cu–ZnO integrated catalysts. It is worth remembering that NH3-TPD interpretation is challenging because it depends on experimental conditions (e.g. amount of sample, pre-treatment) and several desorption phenomena (e.g. NH3 diffusion within pore structure and re-adsorption) [34, 35]. Often peak deconvolution models are needed to fully interpret NH3-TPD profiles. To add to the NH3-TPD interpretation complexity, one must consider that ZSM-5 zeolites can have 96 different Brønsted acid sites (oxygen bridging a Si and a Al atoms in the framework, although two positions are predominantly stable) [36, 37], which would need to be included into the NH3-TPD peak model. Hence, NH3-TPD measurements in this work are discussed in a qualitative manner. Figure S5 shows NH3-TPD profiles for activated ZSM-5 zeolites. Two distinct peaks were observed; a first desorption peak between 230 and 260 °C associated with weakly bonded NH3, likely to correspond to Lewis acid sites, and a second peak between 440 and 500 °C associated with more strongly bound NH3, likely related to Brønsted acid sites [38]. Although, as observed by the clear shoulder on the first desorption peak for ZSM-5(50), this first peak is made of several desorption contributions. As expected, higher Al content in the zeolite resulted in a higher concentration of acid (higher peak area) sites and stronger acidity (peak shift towards higher temperature) [20]. NH3-TPD profiles for CZZ-CVI and CZZ-OG integrated catalysts are shown in Fig. 4a and Fig. 4b, respectively. The NH3-TPD for integrated catalyst shows that the acid sites of parent zeolites have significantly changed after the addition of Cu and ZnO. Instead of the two distinct peaks observed for ZSM-5 indicating the presence of Lewis and Brønsted acid sites, the integrated catalysts showed multiple NH3 desorption processes with an extra feature associated with medium acidity (300–380 °C). Moreover, the concentration of strong acid sites increased for synthesised integrated catalysts compared to parent zeolites, as also reported for other CuZnO-zeolite hybrid systems [38, 39]. The acid properties between CZZ(50 and 80)-CVI and CZZ(50 and 80)-OG are fairly comparable. However, a considerably higher concentration of strong acid sites was detected for CZZ(23)-CVI compared to CZZ(23)-OG.

NH3-TPD profiles for (a) CZZ-CVI and (b) CZZ-OG integrated catalysts over commercial ZSM-5 zeolites with different Si/Al ratios
3.2 Catalyst Performance for DME Production
Synthesised CZZ integrated catalysts are bifunctional catalysts, containing Cu-active sites for the CO2 hydrogenation to CH3OH and acid sites within the zeolite for the consecutive dehydration of CH3OH to DME. The catalytic CO2 hydrogenation (CO2/H2/N2 = 1/3/1, 30 ml min−1) to oxygenates (CH3OH and DME) was assessed at 230–310 °C and 20 bar. Prior to reaction catalysts were pre-reduced in situ with 5% H2/Ar (5% H2/Ar, 220 °C, 2 °C min−1, 1 h). Details on how CO2 conversion, product selectivity and productivities can be found in the supporting information (Equation S3–S11).
The catalytic trend expected for the CO2 hydrogenation to CH3OH with temperature was observed on all synthesised catalysts (Table S2). Increasing the reaction temperature from 230 to 310 °C facilitates CO2 activation, which translates into higher CO2 conversion. However, as dictated by thermodynamics, higher temperature favour the endothermic RGWS instead of the exothermic CH3OH (and DME) synthesis, as observed by the increase in CO selectivity and productivity with temperature [7]. For instance, at 20 bar (CO2/H2 = 1/3) the theoretical CH3OH yield decreases from 10.89 (C mol %) at 230 °C to 1.45 (C mol %) at 300 °C, whilst the CO yield increases from 9.10 to 23.02 (C mol %). When CH3OH is simultaneously dehydrated a DME yield of 20.26 (C mol %) can be achieved at 230 °C, but it is limited to 1.39 (C mol %) at 300 °C [7].
CZZ-CVI catalysts showed poor CO2 conversion (ca. 3% at 250 °C) compared to CZZ-OG (c.a. 14–20% at 250 °C). This could indicate that mass transfer of CO2 is limited over CZZ-CVI, which could be attributed to Cu/ZnO blocking the surface of zeolite particles as suggested by TEM. Whilst CZZ-OG catalysts were formed by Cu/ZnO agglomerates not deposited on the zeolite surface, which resulted in an unobstructed zeolite porous structure. Moreover, no hydrocarbons (HC) were observed over CZZ-CVI catalysts, even at 310 °C. The absence of HC on CZZ-CVI catalysts suggested that either CH3OH cannot diffuse into the ZSM-5 acid sites or that HC formed cannot diffuse out of the zeolite pores. Both scenarios suggest that Cu/ZnO might be blocking the entrance of ZSM-5 channels. The selectivity towards oxygenates over CZZ-OG catalysts is higher than over CZZ-CVI, higher selectivity in addition to higher CO2 conversion translated in almost a six-fold higher oxygenate productivity over CZZ-OG compared to CZZ-CVI at 230 °C (Fig. 5). As expected from thermodynamics, the higher oxygenate productivity was observed at lower temperature, while increasing the reaction temperature above 250 °C resulted in a decrease in oxygenate productivity over CZZ(80 and 50)-OG catalysts. This can be assigned to equilibrium limitations at high temperature, for both CH3OH and DME synthesis from CO2 hydrogenation [7], as well as to the more favoured side reactions, leading to the formation of hydrocarbons [32]. Increasing the Si/Al ratio from 50 to 80 favoured CH3OH dehydration to DME, leading to higher oxygenates productivity. However, further increasing the Si/Al ratio, as in CZZ(23)-OG, resulted in the conversion of oxygenates to undesired by-products.

Oxygenate (CH3OH + DME) productivity obtained over CZZ-OG and CZZ-CVI catalysts (20 bar, CO2/H2/N2 = 1/3/1, 30 ml min−1)
The formation of undesired hydrocarbons is to be avoided not only because it decreases the selectivity towards oxygenates and complicates the separation process, but also because it is normally associated with zeolites deactivation through coke formation [40]. ZSM-5 zeolites are widely used for the synthesis of HC from CH3OH and DME [41,42,43], in the so called methanol-to-hydrocarbons process (MTH) [44]. On MTH, methanol absorbs on Brønsted acid sites yielding surface methoxy species, methoxy species recombine forming C–C bonds (carbene species) with increasing reaction temperature [45]. Carbene species promote the autocatalytic formation of hydrocarbons via the dual-cycle mechanism [46,47,48]. The Si/Al ratio determines the concentration of acid sites on zeolites, increasing the Al concentration results in a higher concentration of Brønsted acid sites, increasing the chance of forming active carbene species leading to greater hydrocarbon formation [49]. The effect of increasing the Al content on CZZ-OG catalysts on the productivity of hydrocarbons is shown in Fig. 6. No HC were detected over CZZ(80)-OG and CZZ(50)-OG below 270 and 250 °C, respectively. HC selectivity remained low (< 0.03%) for CZZ(50 and 80)-OG catalysts at 290 °C, with ethane being the only by-product detected. Nevertheless, a selectivity of c.a. 0.5% towards HC was observed over CZZ(23)-OG, even at 230 °C. Increasing the reaction temperature from 230 to 270 °C resulted in an increase in the HC selectivity to 3.4%. As shown in Fig. 5, oxygenate productivity for CZZ(23)-OG does not increase with temperature up to 250 °C, as observed for CZZ(50 and 80)-OG, but instead decreases steadily with temperature, indicating the conversion of oxygenates to undesired by-products. Over CZZ(23)-OG a range of by-products up to C5 hydrocarbons were detected. Further increasing the reaction temperature to 310 °C resulted in a drop in HC productivity (Table S2), which likely relates to low CH3OH availability.

Oxygenates and hydrocarbons productivity for CZZ-OG integrated catalysts as a function of temperature (20 bar, CO2/H2/N2 = 1/3/1, 30 ml min−1)
4 Conclusions
The preparation methodology of integrated catalysts has a great influence on the Cu/ZnO crystallite size and their distribution over ZSM-5 particles. Thus, influencing the CO2 hydrogenation activity towards CH3OH and the concomitant dehydration to DME.
CVI preparation leads to bigger Cu and ZnO nanoparticles, and an uneven distribution on CZZ-CVI catalysts: small nanoparticles evenly distributed over the surface of ZSM-5 were observed, whilst what appeared to be ZSM-5 areas free of metal showed particle agglomeration under the TEM electron beam. Suggesting that Cu and ZnO were deposited in the outermost zeolite pores or as a film at the surface, leading to poor CO2 conversion and to possibly mass transfer limitations. In this case, possibly a better route to avoid mass transfer limitations would be to prepare Cu/ZnO methanol catalysts by CVI and physically mixed them with ZSM-5 zeolites.
OG resulted in Cu/ZnO agglomerates, made of smaller 20 nm aggregates, that did not obstruct the zeolite pores. Which resulted in higher CO2 conversion and oxygenates productivity compared to CZZ-CVI catalysts. The presence of acid sites is necessary to dehydrate CH3OH to DME. However, a compromise between acid sites concentration is required to avoid excessive formation of hydrocarbons. The high concentration of acid sites on CZZ(23)-OG led to the conversion of oxygenates to HC, whilst no HC were detected for CZZ(50 and 80)-OG below 250 °C, the temperature with the higher oxygenates productivity (ca. 800 mmol kgcat−1 h−1 over CZZ(50)-OG).
References
Schlögl R (2010) The role of chemistry in the energy challenge. Chemsuschem 3:209–222
Carmo M, Fritz DL, Mergel J, Stolten D (2013) A comprehensive review on PEM water electrolysis. Int J Hydrog Energy 38:4901–4934
Wang W, Wang S, Ma X, Gong J (2011) Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 40:3703–3727
Bowker M (2019) Methanol synthesis from CO2 Hydrogenation. ChemCatChem 11:4238–4246
Sheldon D (2017) Methanol production—A technical history. Johns Matthey Technol Rev 61:172–182
Olah GA (2005) Beyond oil and gas: The methanol economy. Angew Chem Int Ed 44:2636–2639
Shen W-J, Jun K-W, Choi H-S, Lee K-W (2000) Thermodynamic investigation of methanol and dimethyl ether synthesis from CO2 Hydrogenation. Korean J Chem Eng 17:210–216
Ruiz Esquius J, Bahruji H, Taylor SH, Bowker M, Hutchings G (2020) CO2 hydrogenation to CH3OH over PdZn catalysts, with reduced CH4 production. ChemCatChem 12:6024–6032
Arcoumanis C, Bae C, Crookes R, Kinoshita E (2008) The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 87:1014–1030
Xu M, Lunsford JH, Goodman DW, Bhattacharyya A (1997) Synthesis of dimethyl ether (DME) from methanol over solid-acid catalysts. Appl Catal Gen 149:289–301
Álvarez A, Bansode A, Urakawa A, Bavykina A, Wezendonk T, Makkee M, Gascon J, Kapteijn F (2017) Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem Rev 117:9804–9838
Spencer MS (1999) The role of zinc oxide in Cu/ZnO catalysts for methanol synthesis and the water–gas shift reaction. Top Catal 8:259
Twigg MV, Spencer MS (2003) Deactivation of copper metal catalysts for methanol decomposition, methanol steam reforming and methanol synthesis. Top Catal 22:191–203
Spencer MS (1987) α-Brass formation in copper/zinc oxide catalysts: I. Bulk equilibrium concentrations of zinc under methanol synthesis and water-gas shift reaction conditions. Surf Sci 192:323–328
Spencer MS (1987) α-brass formation in copper/zinc oxide catalysts: II. Diffusion of zinc in copper and α-brass under reaction conditions. Surf Sci 192:329–335
Spencer MS (1987) α-brass formation in copper/zinc oxide catalysts: III. Surface segregation of zinc in α-brass. Surf Sci 192:336–343
Burch R, Golunski SE, Spencer MS (1990) The role of copper and zinc oxide in methanol synthesis catalysts. J Chem Soc Faraday Trans 86:2683–2691
Aguayo AT, Ereña J, Sierra I, Olazar M, Bilbao J (2005) Deactivation and regeneration of hybrid catalysts in the single-step synthesis of dimethyl ether from syngas and CO2. Int Conf Gas-Fuel 05(106):265–270
Naik SP, Ryu T, Bui V, Miller JD, Drinnan NB, Zmierczak W (2011) Synthesis of DME from CO2/H2 gas mixture. Chem Eng J 167:362–368
Frusteri F, Bonura G, Cannilla C, Drago Ferrante G, Aloise A, Catizzone E, Migliori M, Giordano G (2015) Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation. Appl Catal B Environ 176–177:522–531
Catizzone E, Bonura G, Migliori M, Frusteri F, Giordano G (2018) CO2 Recycling to dimethyl ether: state-of-the-art and perspectives. Molecules 23(1):1420–3049
Munnik P, de Jongh PE, de Jong KP (2015) Recent developments in the synthesis of supported catalysts. Chem Rev 115:6687–6718
Li J-L, Inui T (1996) Characterization of precursors of methanol synthesis catalysts, copper/zinc/aluminum oxides, precipitated at different pHs and temperatures. Appl Catal Gen 137:105–117
Li J-L, Inui T (1996) Enhancement in methanol synthesis activity of a copper/zinc/aluminum oxide catalyst by ultrasonic treatment during the course of the preparation procedure. Appl Catal Gen 139:87–96
Jingfa D, Qi S, Yulong Z, Songying C, Dong W (1996) A novel process for preparation of a Cu/ZnO/Al2O3 ultrafine catalyst for methanol synthesis from CO2 + H2: comparison of various preparation methods. Appl Catal Gen 139:75–85
Ning W, Shen H, Liu H (2001) Study of the effect of preparation method on CuO-ZnO-Al2O3 catalyst. Appl Catal Gen 211:153–157
Forde MM, Kesavan L, Bin Saiman MI, He Q, Dimitratos N, Lopez-Sanchez JA, Jenkins RL, Taylor SH, Kiely CJ, Hutchings GJ (2014) High activity redox catalysts synthesized by chemical vapor impregnation. ACS Nano 8:957–969
Forde MM, Armstrong RD, Hammond C, He Q, Jenkins RL, Kondrat SA, Dimitratos N, Lopez-Sanchez JA, Taylor SH, Willock D, Kiely CJ, Hutchings GJ (2013) Partial oxidation of ethane to oxygenates using Fe- and Cu-containing ZSM-5. J Am Chem Soc 135:11087–11099
Bahruji H, Armstrong RD, Ruiz Esquius J, Jones W, Bowker M, Hutchings GJ (2018) Hydrogenation of CO2 to dimethyl ether over brønsted acidic PdZn catalysts. Ind Eng Chem Res 57:6821–6829
Bahruji H, Bowker M, Jones W, Hayward J, Ruiz Esquius J, Morgan DJ, Hutchings GJ (2017) PdZn catalysts for CO2 hydrogenation to methanol using chemical vapour impregnation (CVI). Faraday Discuss 197:309–324
Sing K (2001) The use of nitrogen adsorption for the characterisation of porous materials. Colloids Surf Physicochem Eng Asp 187–188:3–9
Bateni H, Able C (2019) Development of heterogeneous catalysts for dehydration of methanol to dimethyl ether: A review. Catal Ind 11:7–33
Jiang S, Hwang J-S, Jin T, Cai T, Cho W, Baek YS, Park SE (2004) Dehydration of methanol to dimethyl ether over ZSM-5 zeolite. Bull Korean Chem Soc 25:185–189
Arena F, Dario R, Parmaliana A (1998) A characterization study of the surface acidity of solid catalysts by temperature programmed methods. Appl Catal Gen 170:127–137
Karge HG, Vera D (1990) Investigation of the distribution of acidity in zeolites by temperature-programmed desorption of probe molecules. I Dealuminated mordenites J Phys Chem 94:765–772
Schröder K-P, Sauer J (1993) Siting of Al and bridging hydroxyl groups in zeolite catalysts. Computer simulations of their structure, vibrational spectra and acidity. In: von Ballmoos R, Higgins JB, Treacy MMJ (eds) Proceedings from the Ninth International Zeolite Conference. Butterworth-Heinemann, Oxford, pp 687–694
Brändle M, Sauer J (1998) Acidity differences between inorganic solids induced by their framework structure. A combined quantum mechanics/molecular mechanics ab initio study on zeolites. J Am Chem Soc 120:1556–1570
Bonura G, Cordaro M, Spadaro L, Cannilla C, Arena F, Frustaeri F (2013) Hybrid Cu–ZnO–ZrO2/H-ZSM5 system for the direct synthesis of DME by CO2 hydrogenation. Appl Catal B Environ 140–141:16–24
Bonura G, Migliori M, Frusteri L, Cannilla C, Catizzone E, Giordano G, Frusteri F (2018) Acidity control of zeolite functionality on activity and stability of hybrid catalysts during DME production via CO2 hydrogenation. J CO2 Util 24:398–406
Hwang A, Bhan A (2019) Deactivation of zeolites and zeotypes in methanol-to-hydrocarbons catalysis: Mechanisms and circumvention. Acc Chem Res 52:2647–2656
Rownaghi AA, Hedlund J (2011) Methanol to gasoline-range hydrocarbons: influence of nanocrystal size and mesoporosity on catalytic performance and product distribution of ZSM-5. Ind Eng Chem Res 50:11872–11878
Chang CD, Chu CT-W, Socha RF (1984) Methanol conversion to olefins over ZSM-5: I. Effect of temperature and zeolite SiO2Al2O3. J Catal 86:289–296
Chang CD, Lang William H (1977) Process for manufacturing olefins. Mobil Oil Corporation, US4025576A
Yarulina I, Chowdhury AD, Meirer F, Weckhuysen BM, Gascon J (2018) Recent trends and fundamental insights in the methanol-to-hydrocarbons process. Nat Catal 1:398–411
Wang W, Hunger M (2008) Reactivity of surface alkoxy species on acidic zeolite catalysts. Acc Chem Res 41:895–904
Ilias S, Bhan A (2013) Mechanism of the catalytic conversion of methanol to hydrocarbons. ACS Catal 3:18–31
Ilias S, Bhan A (2012) Tuning the selectivity of methanol-to-hydrocarbons conversion on H-ZSM-5 by co-processing olefin or aromatic compounds. J Catal 290:186–192
Dahl IM, Kolboe S (1993) On the reaction mechanism for propene formation in the MTO reaction over SAPO-34. Catal Lett 20:329–336
Khare R, Liu Z, Han Y, Bhan A (2017) A mechanistic basis for the effect of aluminum content on ethene selectivity in methanol-to-hydrocarbons conversion on HZSM-5. J Catal 348:300–305
Acknowledgements
The authors acknowledge the EPSRC (EP/N010531/1) and the UK Catalysis Hub for funding. Information on the data underpinning the results presented here can be obtained from the Cardiff University data catalogue on request (DOI:10.17035/d.2021.0129557921). This manuscript is dedicated to Michael S. Spencer and in memory of his pioneering work on CH3OH synthesis over Cu/ZnO catalysts.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tariq, A., Ruiz Esquius, J., Davies, T. et al. Combination of Cu/ZnO Methanol Synthesis Catalysts and ZSM-5 Zeolites to Produce Oxygenates from CO2 and H2. Top Catal 64, 965–973 (2021). https://doi.org/10.1007/s11244-021-01447-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-021-01447-8