
Abstract
We have investigated a series of supported and unsupported nickel and cobalt catalysts, principally using neutron vibrational spectroscopy (inelastic neutron scattering, INS). For an alumina supported Ni catalyst we are able to detect hydrogen on the metal for the first time, all previous work has used Raney Ni. For an unsupported Ni foam catalyst, which has similar behaviour to Raney Ni but with a much lower density, the spectra show that there are approximately equal numbers of (100) and (111) sites, in contrast to Raney Ni that shows largely (111) sites. The observation of hydrogen on cobalt catalysts proved to be extremely challenging. In order to generate a cobalt metal surface, reduction in hydrogen at 250–300 °C is required. Lower temperatures result in a largely hydroxylated surface. The spectra show that on Raney Co (and probably also on a Co foam catalyst), hydrogen occupies a threefold hollow site, similar to that found on Co(\(10\bar{1}0\)). The reduced surface is highly reactive: transfers between cells in a high quality glovebox were sufficient to re-hydroxylate the surface.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The Fischer–Tropsch (FT) process is a well-established chemical reaction that utilises synthesis gas (CO and H2), derived from natural gas, coal or biomass, to produce a wide-range of valuable hydrocarbon products [1, 2]. Currently, worldwide fuel and chemical production is based predominantly on crude oil, although this is not sustainable. Fischer–Tropsch synthesis (FTS) could be a viable alternative; however, the FT process’s economic viability is strongly linked to the oil price [3]. The development of the FT process both industrially and academically, will often follow the trail of the cost and availability of crude oil as a result. Currently there is a resurgence of interest in the FT process for synthetic fuel production, because of the decreasing lifetime of crude oil, as well as a combination of other environmental and political factors [1, 3, 4].
The criteria required for an effective Fischer–Tropsch catalyst is to promote both a high capacity of hydrogen adsorption as well as efficiently dissociate adsorbed CO. Several metals are effective FT catalysts such as ruthenium, nickel, iron and cobalt [1, 4, 5]. Of these, ruthenium is in limited supply and thus expensive and nickel catalysts result in large quantities of undesirable methane and can also produce highly toxic nickel carbonyl compounds [1, 4]. (However, nickel is a very efficient hydrogenation catalyst and is widely used industrially for this purpose [6,7,8]). Consequently, only iron and cobalt based catalysts are currently used industrially as FT catalysts [1, 2]. Iron is abundant and is therefore the cheapest of the active FT catalysts [3]. Iron based catalysts have a lower hydrogenation activity in comparison to cobalt and therefore, have a high selectivity towards the production of olefins and oxygenate products with promoters commonly added to further manipulate the product slate [1, 2]. The current demand for an alternative route to produce lower olefins (C2–C4), high value chemical feedstock, free from the use of crude oil, has led to increased interest in the Fischer–Tropsch to olefins (FTO) process, in which iron-based catalysts are ideally suited [9,10,11,12,13]. However, iron-based catalyst also catalyse the water gas shift reaction and exhibit a much more complex structure under FT conditions resulting in uncertainty over the proposed active phase and operation of the catalysts [1,2,3,4].
On the other hand, cobalt catalysts exhibit a lower water–gas shift activity that makes them particularly advantageous for the conversion of natural gas derived syngas which possesses a high H2:CO ratio [1, 2, 4]. As cobalt is a relatively expensive metal, to maximise its use typically the cobalt metal (10–30 wt%) is dispersed on a high surface area stable oxidic support such as alumina, silica or titania. A second metal (0.05–1 wt%), often a noble metal (e.g. Pt, Ru, Re), is commonly added as a promoter to enhance the reduction process and to keep the Co surface ‘clean’ [1, 2, 14]. Cobalt catalysts provide selectivity towards high molecular weight paraffinic products and are generally used as low temperature FT (LTFT) catalysts. High temperature FT (HTFT) results in more methane production [1,2,3,4], as the purpose of the high temperature is to force the formation of a lighter product slate. There has been a shift away from FT processes that can produce petrol (HTFT) towards wax production, which is then hydrocracked to produce diesel [15, 16].
Cobalt based catalysts exhibit a range of desirable properties which have made them an attractive class of catalysts for FT synthesis including a high hydrogenation activity and that they deactivate less rapidly than iron-based catalysts [1, 2, 17, 18]. Cobalt catalysts were utilised in the first commercial FT plants in 1938–1945 in Germany and Japan and today are currently used by Sasol/QP in the Oryx GTL plant in Qatar, Shell in the SMDS plant in Malaysia as well as the Pearl plant in Qatar [1, 3]. Academic and industrial research over the last 20 years has increased the knowledge base of cobalt Fischer–Tropsch synthesis (FTS) substantially by understanding the effect of crystallite size on activity, selectivity and deactivation mechanisms [19,20,21]. However, despite the process being applied industrially for close to a century, the underlying mechanisms which govern the FT reaction remain under debate.
While the interaction of carbon monoxide with cobalt has been extensively studied (e.g. [22,23,24,25]), the state of hydrogen on the surface of cobalt has been much less studied. In particular, studies with vibrational spectroscopy are very rare. This is probably because of the usual reasons: adsorbed hydrogen generally gives weak bands and often they occur in the low energy region of the spectrum that is masked by the intense bands of the support; hydrogen on platinum exemplifies this issue ([26] and references therein). Hydrogen on single crystal Co(\(10\bar{1}0\)) under ultra high vacuum (uhv) has been studied by high resolution electron energy loss spectroscopy (HREELS) [27]. Inelastic neutron scattering (INS) spectroscopy is a form of vibrational spectroscopy that is particularly sensitive to vibrations involving hydrogen motion [25] and has been extensively used to study hydrogen on, and in, catalysts [26, 28,29,30], however, to date, it has been little used for cobalt catalysts [31, 32].
In contrast, the vibrational spectroscopy of hydrogen on nickel has been extensively investigated. HREELS investigations of a variety of surfaces under uhv have been made: (111) [33, 34], (110) [35], (100) [36, 37] and (311) [38] (the work is reviewed in [39]) and there are several INS studies of hydrogen on Raney nickel [40,41,42,43]. As far as we are aware, to date, hydrogen on supported nickel catalysts has not been observed by vibrational spectroscopy.
At the outset of this work, our aim was to use INS spectroscopy to characterise the adsorption sites of hydrogen on a range of unsupported and supported nickel and cobalt catalysts. As we will show, this proved to be much more difficult than we expected. In the bulk of the paper, we describe the trials and tribulations encountered and we conclude with some thoughts as to why these occurred and how future work may overcome them.
2 INS Spectroscopy
INS spectra were recorded with the TOSCA [44, 45] and MAPS [46, 47] spectrometers at the ISIS Neutron and Muon Source [48]. As described in detail elsewhere [28, 46], while the two spectrometers operate over the complete “infrared spectroscopy” range of 0–4000 cm−1, they have different operating principles. Neutrons have mass, consequently INS spectra are a function of both energy transfer (ω, cm−1) and momentum transfer (Q, Å−1). TOSCA follows a fixed trajectory through (Q,ω) space, such that there is a single Q value for each energy transfer value. This means that energy transfer values in the O–H stretch region, ~ 3000–3700 cm−1, are associated with large momentum transfer and the spectra are dominated by multiphonon processes whose intensity varies as Q2n, where n ≥ 2 and the informative fundamental modes are unobservable. This has the result that TOSCA spectra are optimal below 2000 cm−1. In contrast, with MAPS the incident beam (Ei) is a single energy selected from a polychromatic pulse of neutrons. This provides a spectrum in the range 0–Ei, and the resolution is a fraction of Ei (~ 1.5% for MAPS) thus, to obtain good resolution across the entire spectral range, several incident energies are required. Note that improved resolution comes with reduced spectral range. MAPS can access a wide swath of (Q,ω) space in a single measurement, thus by selecting only the small Q data, the high energy fundamentals are observable. Modes at lower energy are seen, but with poorer resolution than with TOSCA.
The complementarity of MAPS and TOSCA is illustrated in Fig. 1. The top part shows the S(Q,ω) map of one of the samples (Co foam as received under 900 mbar hydrogen) recorded with MAPS using Ei = 650 meV (5243 cm−1). The solid black lines are the trajectories of the forward and backscattering detectors on TOSCA (in all the spectra shown here, these are summed and the average is displayed). Figure 1a is the MAPS data shown in the top part, summed over the range 0 ≤ Q ≤ 10 Å−1 (unless otherwise stated, this range was used for all of the MAPS spectra shown). A broad O–H stretch centred at 3540 cm−1 is easily seen, Fig. 1b, however, there is little detail at lower energies. The TOSCA data, Fig. 1c, shows much more detail at lower energy but there is no sign of the O–H stretch. Thus it is the combination of the two types of spectrometer that provides the best description of the chemical system under investigation.

Top: S(Q,ω) map of the Co foam sample as received under 900 mbar hydrogen recorded with MAPS using Ei = 650 meV. The solid black lines show the trajectories through (Q,ω) space of the forward scattering (left trace) and backscattering (right trace) detectors on TOSCA. Bottom: a The MAPS data shown in the top part, summed over the range 0 ≤ Q ≤ 10 Å−1. b × 10 ordinate expansion of the 2800–4000 cm−1 range. Note that the O–H stretch is clearly visible. c The same sample recorded on TOSCA and d × 4 ordinate expansion of the 2800–4000 cm−1 range. Note that the O–H stretch is not visible
3 Experimental
Table 1 lists the materials studied and their source. The Raney Co was supplied by Activated Metal Catalysts as a slurry in water. This was filtered at a Buchner pump and transferred to an aluminium cell. This was all done under a blanket of flowing argon, as Raney Co is potentially pyrophoric in air. For the Ni and Co foam catalysts (prepared under license from Alantum Europe GmbH as described in [49, 50]), a wet sample was transferred into a glass vessel and treated under flowing, pure dry nitrogen (80 l/h) which was constantly removed by a vacuum pump. After 10 min the vacuum was adjusted to 400 mbar. After 20 min the catalyst was heated to 150 °C. After 3 h no more water was seen by visual inspection. After the sample was cooled to room temperature, the vacuum pump was stopped. The sample was flushed for 5 min with flowing nitrogen. The sample was transferred into a glove box and sealed into an INS can and cyclically treated with hydrogen in the range ~ 10−3 mbar to 1220 mbar for 12 h until no further hydrogen consumption/adsorption was measured (capacitive pressure transducer, Baratron). As may be seen from Table 1, as a result of the preparation procedure, all of the unsupported metal catalysts had residual Al bound in various intermetallic phases which could not be leached by alkaline treatment. The Co/C and CoCuMn/C catalysts were prepared as described previously [51, 52].
Pt promoted Co/SiO2 and Co/Al2O3 Fischer–Tropsch catalysts were prepared via the wet impregnation of silica and γ-alumina with solutions of Co(NO3)2·6H2O and [Pt(NH3)4](NO3)2. The total pore volume of the oxide support and maximum solubility of cobalt nitrate in water limit the Co loading that can be achieved in a single impregnation. To obtain a weight loading of ~ 20 wt%, materials were prepared via a double impregnation of the oxidic support with an intermediate calcination step following the first impregnation according to the following methods.
For the Co/Pt on silica catalyst, cobalt (II) nitrate hexahydrate (15.8964 g) and tetraammineplatinum(II) nitrate (1 mL of a 31 mM stock solution) were dissolved in water (25 mL) and stirred on a rotary evaporator at 60 °C for 10 min at atmospheric pressure. The silica support (Davisil Grade 62, pore size 150 Å, pore volume 1.15 cm3/g, 20.0276 g) was then added to the Co solution. A further 15 mL of water was required to form a slurry, which was stirred on the rotary evaporator for a further 10 min. The rotary evaporator pressure was then reduced to 280 mbar for 30 min. The temperature of the water bath was increased to 75 °C for 1.5 h, then to 85 °C for 1 h and finally the pressure was reduced to 50 mbar, in 10 mbar increments, for 3 h. A dark pink powder (32.4097 g) was obtained and calcined at 250 °C (ramp rate 1 °C/min) in a vertical calcination tube for 6 h under a continuous air flow (1000 mL/min) to yield Co3O4–SiO2 (22.7431 g). For the second impregnation, cobalt nitrate (11.4126 g) and tetraammineplatinum(II) nitrate (1.7 mL of the same stock solution) were dissolved in water (35 mL) and stirred on a rotary evaporator at 60 °C for 10 min at atmospheric pressure. Calcined material from the first impregnation (20.0531 g) was added to the Co solution and the mixture was stirred for a further 10 min. The rotary evaporator pressure was then reduced to 280 mbar for 30 min. The temperature of the water bath was increased to 75 °C for 1.5 h, then to 85 °C for 1 h and finally the pressure was reduced to 50 mbar, in 10 mbar increments, for 3 h. The material was calcined in a vertical tube furnace for 6 h under an air flow (1000 mL/min) to yield Co3O4–SiO2 (22.9085 g). The material was characterised by BET (surface area: 205 m2/g, pore size: 134 Å), XRD (Panalytical X’Pert Pro X-ray diffractometer, Co Kα1,2 radiation from 10° to 90° (2θ), scan rate 1.34°/min, at ambient temperature) (Co3O4, 15.5(1) nm) and H2 chemisorption (Micromeritics ASAP 2020) (43 m2/g).
For the Co/Pt on γ-alumina catalyst, cobalt (II) nitrate hexahydrate (15.8921 g) and tetraammineplatinum(II) nitrate (1 mL of a 31 mM stock solution) were dissolved in water (25 mL) and stirred on a rotary evaporator at 60 °C for 10 min at atmospheric pressure. The γ-alumina support (Puralox (Sasol), pore size 127 Å, pore volume 0.5272 cm3/g, 20.0056 g) was then added to the Co solution and stirred on the rotary evaporator for a further 10 min. The rotary evaporator pressure was then reduced to 280 mbar for 30 min. The temperature of the water bath was increased to 75 °C for 1.5 h, then to 85 °C for 1 h and finally the pressure was reduced to 50 mbar, in 10 mbar increments, for 3 h. A dark pink powder (30.8973 g) was obtained and calcined at 250 °C (ramp rate 1 °C/min) in a vertical calcination tube for 6 h under a continuous air flow (1000 mL/min) to yield Co3O4–Al2O3 (23.2340 g). For the second impregnation, cobalt nitrate (11.4121 g) and tetraammineplatinum(II) nitrate (1.7 mL of the same stock solution) were dissolved in water (35 mL) and stirred on a rotary evaporator at 60 °C for 10 min at atmospheric pressure. Calcined material from the first impregnation (20.0132 g) was added to the Co solution and the mixture was stirred for a further 10 min. The rotary evaporator pressure was then reduced to 280 mbar for 30 min. The temperature of the water bath was increased to 75 °C for 1.5 h, then to 85 °C for 1 h and finally the pressure was reduced to 50 mbar, in 10 mbar increments, for 3 h. The material was calcined in a vertical tube furnace for 6 h under an air flow (1000 mL/min) to yield Co3O4–Al2O3 (22.5938 g). The material was characterised by BET (surface area: 95 m2/g, pore size: 103 Å), XRD (Co3O4, 13.7(1) nm) and H2 chemisorption (56 m2/g).
Infrared spectra of the Co3O4–SiO2 and Co3O4–Al2O3 materials were recorded using a Thermo Nicolet Avatar spectrometer (4 cm−1 resolution, 64 scans). The samples (ca 150 mg) were reduced in a custom-built DRIFTS chamber under flowing hydrogen (20 mL/min) at 425 °C (ramp rate of 20 °C/min) for 3 h. Spectra were measured after cooling to room temperature under a hydrogen atmosphere. The sample spectrum was ratioed against a background obtained of dried KBr packed into the DRIFTS chamber.
The catalysts were subjected to a variety of treatments as listed in Table 2. All of these were carried out using a rig [53] designed to handle the large quantities (30–50 g for metal-only catalysts, 5–10 g for supported catalysts) required for INS measurements [28, 29, 46]. Most of the INS spectra were recorded from samples in aluminium cells that are of the same design as the Inconel cells described previously [53], so allow hydrogen to be flowed through them. They are indium-wire sealed and have maximum operating conditions of 1 barg at 120 °C. The Inconel cells have the advantage that they are rated to 500 °C at 20 barg [53], so allow reduction to be carried out at higher temperatures. In some cases, spectra were recorded with the sample in Inconel cell. The advantage of the Al cells is that they have a much lower background than stainless steel or Inconel cells [53]. After activation, transfers between cells were carried out in an argon-filled glove box (MBraun UniLab MB-20-G, [O2], [H2O] < 1 ppm).
4 Results
In the following sections we describe the results obtained for each of the catalysts in turn. As we were unable to source a pure cobalt Raney Co catalyst, we used a Raney Co(81)Ni(16)Al(3) material. As a check on our methodology, the spectrum of hydrogen on nickel analogues of some of the cobalt catalysts used here was also measured. All previous work involving INS studies of hydrogen on nickel have utilised Raney Ni [40,41,42,43], thus the use of supported catalysts is novel and industrially relevant. The studies of the Ni catalysts will be presented first and then the cobalt systems.
4.1 Nickel Catalysts
4.1.1 Ni/Al2O3 and Ni/C
Figure 2 shows the spectra of the supported nickel catalysts. Figure 2a–c shows the Ni/Al2O3 catalyst under hydrogen, after evacuation and the difference spectrum. This shows a broad, weak feature at 940 cm−1 that is assigned to hydrogen on Ni(111) facets [40,41,42,43].

INS spectra of: a Ni/Al2O3 after evacuation at 250 °C and pressurized to 500 mbar with H2, b the same sample after evacuation at 250 °C, c the difference spectrum i.e. (a–b). a and b are on the same ordinate scale, c is × 3 ordinate expanded relative to them. d Ni/C after evacuation at 250 °C and pressurized to 500 mbar with H2, e after evacuation at 200 °C and f the difference spectrum i.e. (d–e). d–f are all on the same scale
In contrast, the only feature for the Ni/C catalyst (Fig. 2d) is a weak mode at 650 cm−1. (The strong features below ~ 400 cm−1 are due to the steel can). The spectrum is unusual in that carbon supports typically give well-defined spectra (e.g. [55,56,57]). This is because the carbon is not present as a standard support material, rather it is the residue from the precursor to controlled decomposition to generate finely divided metal on an unconventional carbon “support” so as to provide good metal dispersion [52]. The difference spectrum shows only a very broad band centred at ~ 600 cm−1 that does not match that expected for hydrogen on nickel. This may be the bending mode of hydroxyls on the Ni, but without MAPS data, this assignment is very tentative.
4.1.2 Ni Foam
The foam catalysts (Ni and Co) are an evolution of the Raney process. In particular, due to their high porosity and low filling weight, substantially less metal mass is required as compared to other activated metal catalyst solutions for fixed-bed applications. The Evonik Metalyst MC™ 9 foam catalysts offer high activity and selectivity, especially for mass transfer limited industrial fixed-bed hydrogenation processes [49, 50].
Figure 3 shows the spectra of the Ni foam sample. In the top of the figure, the features below ~ 400 cm−1 are due to the steel can. From the MAPS spectrum in the O–H stretch region (Fig. 3a), it is apparent that residual hydroxyls are present, although the amount is small and that ~ 50% are lost on evacuation at 200 °C (Fig. 3c). The spectra clearly show the presence of adsorbed hydrogen (broad band from 400 to 1200 cm−1). This is more clearly seen in the difference spectrum, (Fig. 3d). Room temperature evacuation removes the physisorbed H2 (Fig. 3f). The overnight evacuation at 200 °C removes the adsorbed hydrogen (Fig. 3e). There are hints of peaks at 900 and 1030 cm−1, which by comparison to Raney Ni [40,41,42,43], see Fig. 4, are assigned to hydrogen on (111) facets. However, there is also a long tail to low energy, whereas on Raney Ni there were only weak peaks. These are assigned to hydrogen on “non-(111)” facets. The greater intensity of these shows that there is a much larger degree of surface heterogeneity in this sample as compared to Raney Ni. A crude estimation, based on the relative area of hydrogen on the (111) sites to the total area, would indicate that there are approximately equal numbers of (111) and non-(111) sites in the foam catalyst, whereas for Raney Ni the ratio is at least 5:1 (111) to non-(111). We note that there is 10% Al present in the Ni foam catalyst (Table 1), however, this is similar to that found for Raney Ni [58], so probably does not account for the different surface structures of the foam and Raney Ni catalysts.

INS spectra of the Ni foam sample. a Under 1 bar H2, b after evacuation at room temperature to 10 mbar, c after overnight evacuation at 200 °C. d–f Difference INS spectra: d = (under 1 bar H2)—(evacuated at 200 °C) i.e. (a–c), e = —(evacuated to 10 mbar)—(evacuated at 200 °C) i.e.(b–c) and f = (under 1 bar H2)—(evacuated to 10 mbar) i.e. (a–b). In the top part of the figure, the 0–2000 cm−1 range is recorded with TOSCA and the 2400–4000 range is recorded with MAPS. Data from each instrument is plotted on the same scale, (so relative intensities may be compared) but the MAPS and TOSCA data are on different ordinate scales

Difference INS spectrum of hydrogen on: a the Ni foam sample (red). b on Raney Ni (blue) [40] and c positions of Ni–H stretch modes on Ni(110) (Γ-point only, olive bars) [35] and Ni(100) (orange bars, the width of the band centered at ~ 790 cm−1 shows the range that the mode is dispersed across) [36, 37]
HREELS investigations of the high coverage phases of hydrogen on Ni(110) [35] show modes at 637, 871 and 1065 cm−1. On Ni(100) [36, 37] the perpendicular mode (symmetric stretch) is at 645 cm−1 and is essentially dispersionless (i.e. there is no change in transition energy as a function of wavevector), whereas the parallel mode (asymmetric stretch) is strongly dispersed, being at 710 cm−1 at the surface Brillouin zone Γ-point and at 850 cm−1 at the X-point. As INS spectroscopy observes all wavevectors [28], this will result in the mode being spread across this range. As most of the additional intensity (as compared to Raney Ni) occurs in the range 500–850 cm−1, this suggests that the surface of the foam catalyst has a much larger number of (100) facets than Raney Ni, which is largely composed of (111) facets.
4.2 Cobalt Catalysts
In the bulk, cobalt exhibits two polymorphs [59], hexagonally close-packed (hcp) and face-centered cubic (fcc), with the hcp phase transforming to the fcc phase above 422 ± 5 °C [60]. It has been reported that for cobalt particles less than 40 nm, the predominant phase should be fcc [61]. However, the temperature of reduction, addition of promoters and the support influence the relative amounts of the phases. In particular, with alumina the fcc phase is obtained [62, 63], while on silica fcc [64] or a mixture of both phases was obtained [62, 63]. Given the pyrophoric nature of the reduced materials, XRD was not possible with our catalysts. However, given the similarity of the supported catalysts to the literature materials, it is probable that our alumina and silica catalysts are largely fcc cobalt. For the unsupported materials it is less clear; the spectra would suggest that they are hcp cobalt but this is unproven.
4.2.1 Co/SiO2
The original plan was to prepare the sample by reducing the catalyst under flowing hydrogen at 200 °C and cool under a hydrogen flow. This would be measured on TOSCA. The sample would then be dehydrogenated and measured again to generate a background. Unfortunately, due to a breakdown in the proton accelerator used for neutron production [48], it was not possible to measure the sample immediately after hydrogenation and it was several months before it could be measured on TOSCA. As shown in Fig. 5a and d, the spectrum is dominated by water (libration at 600 cm−1 and bend at 1660 cm−1) and hydroxyls (bend at 1100 cm−1 and O–H stretch at 3340 cm−1) [65,66,67], suggesting either a slow reaction with the support via spillover or, more likely, that the can had leaked over time. Following this, the sample was reduced and re-hydrogenated again and re-measured giving a clean result, shown in Fig. 5b and e. The sample was then dehydrogenated for the background measurement shown in Fig. 5c.

Spectra of Co/SiO2. (a–e) INS spectra: a original hydrogenated sample, b after reduction and re-hydrogenation, c after evacuation at 200 °C. d and e Difference INS spectra: d = (original hydrogenated sample)—(evacuated at 200 °C) i.e. a–c and e = (reduced and re-hydrogenated)—(evacuated at 200 °C) i.e. (b–c). Note difference in the ordinate-scale compared to (a–d). f Infrared spectrum of a reduced sample. The inset is × 4 ordinate expansion of the 3000–4000 cm−1 region
The difference spectrum of the re-hydrogenated sample, Fig. 5e, shows a very broad band centred at 500 cm−1, with maxima at 320, 465, 675 cm−1. From the literature on Co–H vibrations using HREELS, only the feature at 675 cm−1 is vaguely consistent [27] with the assignment to Co–H. It is possible that the modes are Co–H related or it is also possible they are due to hydroxyls on the cobalt or on the catalyst support. We will return to this point in the Discussion.
The infrared spectrum of the reduced catalyst, Fig. 5f, shows the presence of isolated (sharp band at 3740 cm−1) and hydrogen-bonded hydroxyls (broad band at 3365–3720 cm−1), both on the silica. The spectrum also illustrates an advantage of INS spectroscopy for the study of supported catalysts: the region below 2000 cm−1 is dominated by Si–O stretch and bend modes rendering observation of species on the metal very difficult. In contrast, INS spectroscopy can access the entire 0–4000 cm−1 range, albeit with inferior resolution. The complementarity of the techniques is obvious.
4.2.2 Raney Co: 1st Sample
Figure 6 shows the succession of spectra acquired for the Raney Co catalyst with 650 and 300 meV incident energy on MAPS after subtraction of the empty can. If it was apparent from the 650 meV spectrum that the treatment had not resulted in any change to the spectrum, to save time the 300 meV spectrum was not recorded.

MAPS INS spectra of Raney Co after subtraction of the empty can: a after drying at 100 °C, b dosed with H2, c reduced and dosed with H2, d 1 after 6 h under flowing H2 at 100 °C, e after 250 °C reduction and f reference spectrum of water. The top set of spectra is with 650 meV and the lower with 300 meV incident energy. In each set, the individual spectra (except for f) are plotted on the same ordinate scale so the intensities are directly comparable
It is apparent from Fig. 6 that only the 250 °C reduction made any significant difference to the catalyst. Drying it at 100 °C, irrespective of whether helium or hydrogen was used, resulted in a hydroxylated surface. Figure 7 shows this in more detail. In the O–H stretch region, there is a sharp peak at 3630 cm−1 and a broad one at ~ 3400 cm−1; in the bending region there is an intense, broad peak at 850 cm−1 and a weaker peak at 1700 cm−1. The peaks are all assigned as surface hydroxyl modes: O–H stretch, Co–O–H bend and its first overtone respectively. The only alternative assignment is to adsorbed water. The 3630 cm−1 mode is too high to be water and agrees with that found by HREELS for hydroxyls on Co3O4(111) [68]. As seen in Fig. 6, the 3400 cm−1 peak is close to that found for ice, its larger transition energy would require it to be more weakly hydrogen bonded than water. This assignment would also require that that at least some of the 850 cm−1 band is due to the librational modes of water, which are more usually found to peak at 600–700 cm−1 [69], and crucially, that the 1700 cm−1 was the H–O–H bending mode. 1700 cm−1 would be unusually high for the water bending mode, which is usually found at 1600–1650 cm−1 [69]. Examination of the momentum transfer, (Q, Å−1) enables this possibility to be eliminated [70]. Figure 8a shows the Q-dependence for the 800 cm−1 mode, 8b that of the 1700 cm−1 feature and 8c that of the bending mode of water. It is apparent that the 1700 cm−1 mode peaks at a larger value of Q (Qmax) than either of the other two modes, thus it cannot be the water bending mode.

INS spectra of Raney cobalt after: a drying, b 250 °C reduction and c reference spectrum of water. The top set of spectra is with 650 meV and the lower with 300 meV incident energy. The vertical bars in the lower set are the energies at which the Co–H modes are found for the high coverage phase of hydrogen on Co \(\left( {10\bar{1}0} \right)\) [27]. In both sets of spectra (a) and (b) are plotted on the same ordinate scale so the intensities are directly comparable

Momentum transfer (Q, Å−1) dependence of: a the 800 cm−1 and b the 1700 cm−1 of dried Raney Co and c the bending mode of water
Further, its Qmax = of 11.8 Å−1 is ~ √2 larger than Qmax = 8.9 of the 800 cm−1 mode, consistent with it being the first overtone of the mode. Thus both O–H stretch modes are assigned to hydroxyls. The difference in transition energy would indicate two populations; one possibility is isolated hydroxyls at 3640 cm−1 and hydrogen-bonded hydroxyls at 3400 cm−1.
After reduction in H2 at 250 °C, the spectrum has dramatically changed, Fig. 7b. The amount of hydroxyls has been reduced by ~ 60%, although there still appear to be two populations present. The O–H stretch for one of these matches that of ice, however, the lower energy modes are not consistent with this. In the low energy region three weak bands are seen at 910, 1140 and 1690 cm−1. There must be a Co–O–H bending mode present, it could be one of the three seen here, or it could be hidden by the tail of the peak at 200 cm−1.
4.2.3 Co/Al2O3
Figure 9 shows the spectra for the Co/Al2O3 sample. The infrared spectrum after reduction, Fig. 9a, shows three peaks in the O–H stretch region at 3728, 3668 and 3587 cm−1. Again, the region below 1200 cm−1 is inaccessible because of adsorption by the support. The INS spectra Fig. 9b, c, d closely resembles that seen for activated η-Al2O3 [71]: O–H stretch at 3720 cm−1 and Al–O–H bend at 890 cm−1. Hence the additional peak at 420 cm−1, together with the tail at 3550 cm−1, must be assigned to hydroxyls on the cobalt nanoparticles. The interaction with H2 results in new bands at 640, 870, 1610 and 3290 cm−1. Subtraction of the spectrum of the reduced catalyst from that of the H2 dosed sample, gives the spectrum shown in Fig. 9d. Comparison with that of ice, Fig. 9e, shows that this is water. That the subtraction works so well in the O–H stretch region, shows that the interaction is with the hydroxyls on the cobalt, as evidenced by the disappearance of the band at 420 cm−1.

Spectra of Co/Al2O3. a Infrared spectrum of the reduced catalyst. The inset is × 6 ordinate expansion of the 3000–4000 cm−1 region. b–f INS spectra with the empty can subtracted: b after reduction, c after second reduction, d after third reduction, e difference spectrum: d–b and f reference spectrum of water. The middle panel is with 650 meV and the lower panel with 300 meV incident energy. In each set, the individual spectra (except for f) are plotted on the same ordinate scale so the intensities are directly comparable
4.2.4 Co/C
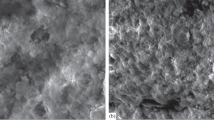
The Co/C and CoCuMn/C (see §2.2.5) catalyst materials are characterized by high metal loading with the metal nanoparticles finely dispersed in a non-graphitic carbon matrix. This is readily apparent from the TEM and the particle size distribution, Fig. 10, that shows the carbon to be highly disordered and most of the particles below 28 nm. Cobalt particles larger than 98 nm are not considered to be significant as there is less than 3% of them. The slightly higher ratios of particles in the intervals 140–150 nm and 42–56 diameter particles are most likely caused by clustering/agglomeration effects; TEM results do not indicate differing types of Co particles. The larger particles and their corresponding small surface area will not contribute significantly to the spectra.

Top: HR-TEM image of Co/C catalyst as prepared, bottom: Co particle size distribution as determined by computer aided analysis of the TEM image
Figure 11a–c show the spectra of the Co/C sample. It can be seen that all three spectra are very similar apart from the intense peak at 46 cm−1. As with the Ni/C catalyst (see §2.1.1) and consistent with the TEM, what is striking is that the spectra do not resemble that of a typical carbon support [55,56,57] or that of graphite, Fig. 11d [72]. The only evidence for the carbon is a very weak C–H stretch mode at 2950 cm−1. This is characteristic of sp3 C–H rather than the sp2 C–H expected for hydrogen-terminated graphene-like sheets (> 3000 cm−1) [73]. The “as received” sample also shows a weak peak at 3490 cm−1 assigned to residual hydroxyls. The low intensity shows that there are very few of these and they are largely removed on evacuation at 200 °C.

INS spectra of the Co/C sample: a original hydrogenated sample, b after evacuation to 10 mbar, c after overnight evacuation at 200 °C and d reference spectrum of graphite [54]. e–g Difference INS spectra: d = (original hydrogenated sample)—(evacuated at 200 °C) i.e. a–c, e = (evacuated to 10 mbar)—(evacuated at 200 °C) i.e. b–c and g = (original hydrogenated sample)—(evacuated to 10 mbar) i.e. (a–b). In the top part of the figure, the 0–2000 cm−1 range is recorded with TOSCA and the 2400–4000 range is recorded with MAPS. Data from TOSCA (except d) are plotted on the same scale, the MAPS data in (c) is × 10 ordinate expansion relative to that in (a). The MAPS and TOSCA data are on different scales
The difference spectra, Fig. 11e–g, show the effects of evacuation more clearly. Subtracting either of the evacuated spectra gives very similar results: there is an intense peak at 46 cm−1, a very broad (> 200 cm−1) feature maximising at ~ 200 cm−1 and a weak, sharp peak at 116 cm−1. The last of these is almost exactly at the transition energy of the of the J = 0 → 1 rotational transition of free H2 and is the same energy as seen for H2 physisorbed on graphite [74]. Accordingly it is assigned to H2 on the support and the broad feature maximising at 200 cm−1 is characteristic of H2 recoil [75]. The intense band at 46 cm−1 is interesting. Two possibilities are: (i) it is the Co–O–H out-of-plane bending mode, (ii) it is H2 on Co. The first of these appears unlikely as the O–H stretch is very weak and this mode has not been seen on any of the other cobalt catalysts. In particular, the Co/SiO2 and Co/Al2O3 spectra suggest that the Co–O–H bending mode occurs around 400 cm−1 and there is no evidence for this. The second possibility is both intriguing and highly speculative. Examples of H2 coordinated to cations in zeolites (Ca exchanged Na-Y with J = 0 → 1 rotational transitions at 32.3 and 45.3 cm−1 [76] and Co exchanged AlPO with J = 0 → 1 rotational transition at 58.4 cm−1 [77]) are known that give energies as low as this but not for metals. For the J = 0 → 1 rotational transition to fall to 46 cm−1 from 118 cm−1 would require the H–H bondlength to increase to ~ 1.2 Å from 0.746 Å. Such bondlengths are known for Kubas-type dihydrogen complexes [78]. This implies a strong interaction between the Co nanocluster and H2 that does not dissociate it.
4.2.5 CoCuMn/C
Figure 12 shows the spectra of the CoCuMn/C catalyst. As with the Ni/C and Co/C catalysts there is no indication of the carbon support, in either the C–H deformation or stretch regions. A weak O–H stretch mode is observed at 3590 cm−1 and an intense, broad feature centred at 800 cm−1 with its first overtone at 1650 cm−1. The difference spectrum highlights this more clearly.

INS spectra of the CoCuMn/C sample: original hydrogenated sample recorded with a MAPS (the inset is a × 6 ordinate expansion of the 2800–4000 cm−1 region) and b TOSCA, c after overnight evacuation at 200 °C and d = (original hydrogenated sample)—(evacuated at 200 °C) i.e. (a–c). b, c and d are all plotted on the same ordinate scale
4.2.6 Co Foam
Figure 13 shows the difference spectra for the Co foam sample. From Fig. 13a it is apparent that the adsorbed species is disordered water (librational modes at ~ 800 cm−1, water scissors at 1650 cm−1,

Difference INS spectra of the Co foam sample. a = (original hydrogenated sample)—(evacuated at 200 °C) recorded on MAPS, b same as a except recorded with TOSCA and c = (re-hydrogenated sample)—(evacuated at 200 °C) recorded on MAPS. c is × 14 ordinate expanded relative to (a)
O–H stretch at 3530 cm−1), the corresponding TOSCA spectra, Fig. 13b, show the librational modes and also the translational modes (peak at 49 cm−1). The sharp peak at 49 cm−1 is at a similar position to that seen for the Co/C sample, Fig. 11e, f, g, however, both the shape and position are different and, crucially, there are no additional features in Fig. 11f, whereas there are for the Co foam sample. Hence we assign the two features to different species. This again emphasises the reactivity of an activated cobalt surface.
Re-hydrogenation of the sample after evacuation at 200 °C generates the difference spectrum shown in Fig. 13c. This shows an extremely weak, broad band that stretches from 500 to 1600 cm−1, the weakness is undoubtedly a reflection of the low surface area of the material. However, there are no hydroxyls generated by the addition of hydrogen and the most reasonable assignment is to surface hydrogen. The considerable width of the band is probably a reflection of what is likely to be a highly disordered surface, as was found for the Ni foam sample (see Sect. 4.1.2).
4.2.7 Raney Co: 2nd Sample
Our experience with the Raney Co sample was that a high temperature reduction was required in order to remove the oxide. Accordingly, a second sample was dried and then reduced at 300 °C. This sample was then measured in the Inconel cell and subsequently pressurized to 1 bar H2. Figure 14 shows the results. The difference spectrum for the 650 meV data shows a broad band centred at ~ 880 cm−1, that is more clearly seen in the 300 meV data. Crucially, the 650 meV data shows nothing in the O–H stretch region, so the feature cannot be hydroxyls. Comparison with the transition energies found for the high coverage phase of hydrogen on Co \(\left( {10\bar{1}0} \right)\) [27] shows that the envelope encompasses the modes and that there are sub-maxima at, or close to, the Co–H modes found for Co \(\left( {10\bar{1}0} \right)\).

INS spectra of the second Raney Co sample, all spectra recorded on MAPS. Top: Ei = 650 meV, bottom: Ei = 300 meV. (a) Dried, reduced sample, (b) sample plus 1 bar H2 and c the difference spectrum (a–b). The vertical bars in the lower part are the transition energies at which the Co–H modes are found for the high coverage phase of hydrogen on Co \(\left( {10\bar{1}0} \right)\) at the Brillouin zone Γ-point [27]
5 Discussion
As stated in the Introduction, the aim was to characterise the adsorption sites of hydrogen on a range of nickel and cobalt catalysts. The work with the nickel catalysts, demonstrates that our methodology is reliable as we were able to observe adsorbed hydrogen on a supported Ni catalyst, which to our knowledge is the first time this has been achieved; all previous work has used Raney Ni [40,41,42,43]. The novel Ni foam catalyst is clearly much more heterogeneous in terms of surface sites than Raney nickel as judged by the much larger number of non-(111) sites.
The supported cobalt catalysts demonstrate the reactivity of the hydrogenated surface in that even though the transfers between sample cans was carried out in a high quality argon-filled glove box ([O2], [H2O] < 1 ppm), the spectra consistently showed the presence of water and hydroxyls (Figs. 6, 9, 13).
The Co/C catalyst is intriguing because of the presence of the band at 46 cm−1, the only credible assignment for this, is that it is H2 coordinated to Co. While Kubas-type dihydrogen complexes have modes at this energy, the additional modes expected for complexed dihydrogen are not seen here. We did not observe it on all the catalysts. However, the mode is best seen with TOSCA (and similar instruments) as this region is routinely recorded with this type of spectrometer and TOSCA was not used for all the samples. MAPS is capable of observing transitions at low energy, but it requires the use of Ei < 100 meV. With the higher energies used here (250 and 650 meV), the feature is hidden by the elastic line. It is noteworthy that the Co/C was not transferred between cans.
Our initial treatment of Raney Co was based on our past experience with Raney Ni [43], where temperatures above 100 °C are generally avoided in order to prevent sintering. From Fig. 6a–c, drying in either He or H2 at 100 °C removes water but leaves a hydroxylated surface. Only when reduction is carried out in flowing H2 at 250 °C or above are the hydroxyls significantly reduced, although they are not eliminated. This is in agreement with a recent operando XANES study of a Co–Ni–Re/γ-Al2O3 catalyst [79] that found that reduction from Co3O4 to CoO started at 260 ± 10 °C and the subsequent reduction from CoO to Co(0) occurred above 340 ± 10 °C.
The spectrum of Raney Co after high temperature reduction, shows weak features in the region where the Co–H modes are found for the high coverage phase of hydrogen on Co \(\left( {10\bar{1}0} \right)\) [27]. Based on this experience, we repeated the measurements with Raney Co but with much more stringent conditions: reduction at 300 °C. The INS spectrum was measured and the cell pressurised to 1 bar H2. All of the operations were carried out in an Inconel can, which is why the background in Fig. 14 is so high. The difference spectra show a broad band centred at ~ 800 cm−1, but crucially, there is no O–H stretch present in the 650 meV data. This means that the band cannot be due to water or hydroxyls and instead is unambiguously assigned to hydrogen on the cobalt surface. The sub-maxima agree very well with the transition energies for H on Co\(\left( {10\bar{1}0} \right)\); the width of the feature can be ascribed to the greater heterogeneity of the Raney Co(81)Ni(16)Al(3) surface, coupled with the presence of vibrational dispersion in the modes, as is commonly found for adsorbed hydrogen on metal surfaces: e.g. H on Pt black [80], H on Ni(111) [81], H on W(100) [82]. Table 3 lists the modes observed and their assignments.
The stability of the hydroxyls on Raney Co has implications for the only previous INS study of hydrogen on Raney Co by Chojecki et al. [31]. Figure 15 shows their results. Overlaid on the lower part of the figure are the 300 meV spectra of our first Raney Co sample after drying at 100 °C (Fig. 7c).

In [31], the spectrum after adsorption of H2 was assigned to a variety of adsorption sites based on calculations of small cobalt clusters (four or five Co atoms, peak positions indicated). We note that these are unlikely to be a good model of an extended metal surface. However, the broad underlying peak exactly matches what we see in the dried Raney Co and that has been unequivocally assigned to hydroxyls (Fig. 7a–c). The spectra suggest that Chojecki et al.’s activation conditions (473 K for 6 h in vacuum) resulted in a partially dehydroxylated surface that was immediately re-hydroxylated on exposure to hydrogen. The upshift of the ~ 800 cm−1 band in Fig. 15a to ~ 900 cm−1 in Fig. 15b is consistent with increased hydrogen bonding being present.
6 Conclusions
There are several lessons to be learned from this work. The first is that an oxidic and/or hydroxylated Co surface is very resistant to reduction. An extended period of reduction at > 250 °C is required to remove most of the hydroxyls. It is also clear that the clean surface is highly reactive; our work suggests that even very small amounts of oxygen results in hydroxylation of the surface. This means that transfers between cells, even in a good quality glove box, should be avoided.
It is striking that we were only unambiguously successful in seeing Co–H modes on the unsupported materials. On the supported catalysts we only saw hydroxyls and it is tempting to speculate that this is because of facile spillover on the supported systems that is not possible on the unsupported ones. This is clearly highly contentious because the sample handling (reduction conditions, transfers between cans) is critical and it is quite possible that we have not yet found the “sweet spot” for the supported Co catalysts.
More generally, this work shows that for catalysts that may have hydroxyls present, it is essential to use direct geometry INS spectrometers to check for the presence of O–H stretch modes. Their presence or absence allows a clear-cut distinction to be made between hydroxyls and surface hydrides. There are several examples in the literature where the analysis is predicated on the absence of O–H stretch modes in systems measured by TOSCA-like instruments. Figure 1 graphically demonstrates that this is not a valid assumption: “absence of evidence is not evidence of absence”.
References
van de Loosdrecht J, Botes FG, Ciobica M, Ferreira A, Gibson P, Moodley DJ, Saib AM, Visage JL, Weststrate CJ, Niemantsverdriet JW (2013) Fischer-Tropsch synthesis: catalysts and chemistry. In: Reedijk J, Poeppelmeier K (eds) Comprehensive inorganic chemistry II, vol 7. Elsevier, Oxford, pp 525–557
Steynberg AP (2004) Introduction to Fischer Tropsch technology. Stud Surf Sci Catal 152:1–63
Dry ME (2002) The Fischer-Tropsch process: 1950-2000. Catal Today 71:227–241
Schulz H (1999) Short history and present trends of Fischer-Tropsch synthesis. App Catal A: Gen 186:3–12
Jahangiri H, Bennett J, Mahjoubi P, Wilson K, Gu S (2014) A review of advanced catalyst development for Fischer-Tropsch synthesis of hydrocarbons from biomass derived syn-gas. Catal Sci Technol 4:2210–2229
Lloyd L (2011) Hydrogenation catalysts. In: Handbook of industrial catalysts. Fundamental and applied catalysis. Springer, Boston. https://doi.org/10.1007/978-0-387-49962-8_3
Dijkstra AJ (2012) Kinetics and mechanism of the hydrogenation process—the state of the art. Eur J Lipid Sci Technol 114:985–998
Stoffels MA, Klauck FJR, Hamadi T, Glorius F, Leker J (2020) Technology trends of catalysts in hydrogenation reactions: a patent landscape analysis. Adv Synth Catal 362:1258–1274
Davidson AL, Gibson EK, Cibin G, van Rensburg H, Parker SF, Webb PB, Lennon D (2020) The application of inelastic neutron scattering to investigate iron-based Fischer-Tropsch to olefins catalysis. J Catal 392:197–208
Botes GF, Bromfield TC, Coetzer RIJ, Crous R, Gibson P, Ferreira AC (2016) Development of a chemical selective Fischer Tropsch catalyst. Catal Today 275:40–48
Yuan Y, Huang S, Wang H, Wang Y, Wang J, Lv J, Li Z, Ma X (2017) Monodisperse nano-Fe3O4 on α-Al2O3 catalysts for Fischer-Tropsch synthesis to lower olefins: promoter and size effects. ChemCatChem 9:3144–3152
Galvis HMT, Koeken ACJ, Bitter JH, Davidian T, Ruitenbeek M, Dugulan AI, de Jong KP (2013) Effects of sodium and sulfur on catalytic performance of supported iron catalysts for the Fischer-Tropsch synthesis of lower olefins. J Catal 303:22–30
Paalanen PP, van Vreeswijk SH, Weckhuysen BM (2020) Combined in situ x-ray powder diffractometry/Raman spectroscopy of iron carbide and carbon species evolution in Fe(-Na-S)/α-Al2O3 catalysts during Fischer-Tropsch synthesis. ACS Catal 10:9837–9855
Herbert JJ, Senecal P, Martin DJ, Bras W, Beaumont SK, Beale AM (2016) X-ray spectroscopic and scattering methods applied to the characterisation of cobalt-based Fischer-Tropsch synthesis catalysts. Catal Sci Technol 6:5573–5791
Bouchy C, Hastoy G, Guillon E, Martens JA (2009) Fischer-Tropsch waxes upgrading via hydrocracking and selective hydroisomerization. Oil Gas Sci Technol Rev IFP 64:91–112
Tan Y, Hu W, Du Y, Jiusheng L (2020) Species and impacts of metal sites over bifunctional catalyst on long chain n-alkane hydroisomerization: a review. App Catal A: Gen. https://doi.org/10.1016/j.apcata.2020.117916
O’Brien RJ, Xu L, Spicer RL, Bao S, Milburn DR, Davis BH (1997) Activity and selectivity of iron Fischer-Tropsch catalysts. Catal Today 36:325–334
Jacobs G, Patterson PM, Zhang Y, Das T, Li J, Davis BH (2002) Fischer-Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl Catal A: Gen 233:263–281
Keyvanloo K, Fisher ML, Hecker WC, Lancee RJ, Jacobs G, Bartholomew CH (2015) Kinetics of deactivation by carbon of a cobalt Fischer-Tropsch catalyst: effects of CO and H2 partial pressures. J Catal 327:33–47
Mejía CH, van Deelen TW, de Jong KP (2018) Activity enhancement of cobalt catalysts by tuning metal-support interactions. Nat Commun 9:4459
Iglesia E, Reyes SC, Madon RJ, Soled SL (1993) Selectivity control and catalyst design in the Fischer-Tropsch synthesis: sites, pellets and reactors. Adv Catal 39:221–302
Todorova S, Zhelyazkov V, Kadinov G (1996) IR, TPR and chemisorption study of alumina-supported cobalt catalysts. React Kinet Catal Lett 57:105–110
Beitel GA, de Groot CPM, Oosterbeek H, Wilson JH (1997) A combined in situ PM-RAIRS and kinetic study of single-crystal cobalt catalysts under synthesis gas at pressures up to 300 mbar. J Phys Chem B 101:4035–4043
Khodakov A, Ducreux O, Lynch J, Rebours B, Chaumette P (1999) Structural modification of cobalt catalysts: effect of wetting studied by X-ray and infrared techniques. Oil Gas Sci Technol Rev IFP 54:525–536
Lyu S, Wang L, Zhang J, Liu C, Sun J, Peng B, Wang Y, Rappé KG, Zhang Y, Li J, Nie L (2018) Role of active phase in Fischer-Tropsch synthesis: experimental evidence of CO activation over single-phase cobalt catalysts. ACS Catal 8:7787–7798
Parker SF, Mukhopadhyay S, Jiménez-Ruiz M, Albers PW (2019) Adsorbed states of hydrogen on platinum: a new perspective. Chem Eur J 25:6496–6499
Ernst K-H, Schwarz E, Christmann K (1994) The interaction of hydrogen with a cobalt (\(10\bar{1}0\)) surface. J Chem Phys 101:5388–5401
Mitchell PCH, Parker SF, Ramirez-Cuesta AJ, Tomkinson J (2005) Vibrational spectroscopy with neutrons, with applications in chemistry, biology, materials science and catalysis. World Scientific, Singapore
Albers PW, Lennon D, Parker SF (2017) In: Fernandez-Alonso F, Price DL (eds) Neutron scattering: applications in biology, chemistry, and materials science, Ch. 5. Academic Press, New York, pp 279–348
Polo-Garzon F, Luo S, Cheng Y, Page KL, Ramirez-Cuesta AJ, Britt PF, Wu Z (2019) Neutron scattering investigations of hydride species in heterogeneous catalysis. ChemSusChem 12:93–103
Chojecki A, Jobic H, Jentys A, Müller TE, Lercher JA (2004) Inelastic neutron scattering of hydrogen and butyronitrile adsorbed on Raney-Co catalysts. Catal Lett 97:155–162
Schärringer P, Müller TE, Jentys A, Lercher JA (2009) Identification of reaction intermediates during hydrogenation of CD3CN on Raney-Co. J Catal 263:34–41
Yanagita H, Sakai J, Aruga T, Takagi N, Nishijima M (1997) Adsorbed states of H on N(111) at 100 K: a vibrational study. Phys. Rev. B 56:14952
Okuyama H, Ueda T, Aruga T, Nishijima M (2001) Overtones of H vibrations at N(111): formation of delocalized states. Phys Rev B 63:
Voigtländer B, Lehwald S, Ibach H (1989) Hydrogen adsorption and the adsorbate-induced Ni(110) reconstruction- an EELS study. Surf Sci 208:113–135
Okuyama H, Hossain MZ, Aruga T, Nishijima M (2002) Adsorbate phonons on Ni(100)(1 × 1)-H. Phys Rev B 66:
Ibach H (2020) Quantum motion of hydrogen on Ni(100) surfaces. Phys Rev B 102:
Schilbe P, Siebentritt S, Pues R, Rieder K-H (1996) Adsorption of hydrogen and of oxygen on an open metal surface- HREELS investigation at Ni(311). Surf Sci 360:157–170
Nishijima M, Okuyama H, Takagi N, Aruga T, Brenig W (2005) Quantum delocalization of hydrogen on metal surfaces. Surf Sci Rep 57:113–156
Cavanagh RR, Kelley RD, Rush JJ (1982) Neutron vibrational spectroscopy of hydrogen and deuterium on Raney nickel. J Chem Phys 77:1540–1547
Jobic H, Renouprez A (1984) Inelastic neutron scattering spectroscopy of hydrogen adsorbed on Raney nickel. J Chem Soc Faraday Trans I 80:1991–1997
Jobic H, Clugnet G, Renouprez A (1987) Neutron inelastic spectroscopy of hydrogen adsorbed at different pressures on a Raney nickel catalyst. J Electron Spectrosc Relat Phenom 45:281–290
Parker SF, Bowron DT, Imberti S, Soper AK, Refson K, Lox ES, Lopez M, Albers P (2010) Structure determination of adsorbed hydrogen on a real catalyst. Chem Commun 46:2959–2961
Parker SF, Fernandez-Alonso F, Ramirez-Cuesta AJ, Tomkinson J, Rudić S, Pinna RS, Gorini G, Fernández Castañon J (2014) Recent and future developments on TOSCA at ISIS. J Phys: Conf Ser 554:
Pinna RS, Rudić S, Parker SF, Armstrong J, Zanetti M, Škoro G, Waller SP, Zacek D, Smith CA, Capstick MJ, McPhail DJ, Pooley DE, Howells GD, Gorini G, Fernandez-Alonso F (2018) The neutron guide upgrade of the TOSCA spectrometer. Nucl Inst Methods Phys Res A 896:68–74
Parker SF, Lennon D, Albers PW (2011) Vibrational spectroscopy with neutrons: a review of new directions. Appl Spectrosc 65:1325–1341
Ewings RA, Stewart JR, Perring TG, Bewley RI, Le MD, Raspino D, Pooley DE, Škoro G, Waller SP, Zacek D, Smith CA, Riehl-Shaw RC (2019) Upgrade to the MAPS neutron spectrometer. Rev Sci Instrum 90:
ISIS Neutron and Muon Source. https://www.isis.stfc.ac.uk/Pages/home.aspx. Accessed 18 Oct 2020
Radivojevic D, Naumann D, Saberi S, Bae J, Poss R (2017) Surface modified metallic foam body, process for its production and use thereof. European patent specification EP 2 764 916 B1
Berweiler M, Göttlinger M, Roos M, Schwarz M, Poss R (2018) Process and catalyst for preparing 1,4-butanediol. International patent WO 2018/060269 Al
Wiesmann T, Reinsdorf A, Lohmann H, Wolf D (2020) Development of Co-Cu-C-Mn catalysts for the synthesis of higher alcohols. Chem Ing Tech 92:1459–1466
Chamski S, Kadyrov R, Reinsdorf A, Wolf D (2019) Materials comprising carbon-embedded cobalt nanoparticles, processes for their manufacture, and use as heterogeneous catalyst, Patent application EP 19195500.4
Warringham R, Bellaire D, Parker SF, Taylor J, Goodway CM, Kibble M, Wakefield SR, Jura M, Dudman MP, Tooze RP, Webb PB, Lennon D (2014) Sample environment issues relevant to the acquisition of inelastic neutron scattering measurements of heterogeneous catalyst samples. J Phys: Conf Ser 554:
https://catalysts.evonik.com/product/catalysts/en/pages/article.aspx?articleId=55396. Accessed 18 Oct 2020
Albers PW, Pietsch J, Krauter J, Parker SF (2003) Investigations of activated carbon catalyst supports from different natural sources. Phys Chem Phys 5:1941–1949
Carosso M, Lazzarini A, Piovano A, Pellegrini R, Morandi S, Manzoli M, Vitillo JG, Jimenez Ruiz M, Lamberti C, Groppo E (2018) Looking for the active hydrogen species in a 5 wt% Pt/C catalyst: a challenge for inelastic neutron scattering. Faraday Discuss 208:227–242
Parker SF, Walker HC, Callear SK, Grünewald E, Petzold T, Wolf D, Möbus K, Adam J, Wieland SD, Jimenez Ruiz M, Albers PW (2019) The effect of particle size, morphology and support on the formation of palladium hydride in commercial catalysts. Chem Sci 10:480–489
Lee GD, Suh CS, Park JH, Park SS, Hong SS (2005) Raney Ni catalysts derived from different alloy precursors (i) morphology and characterization. Korean J Chem Eng 22:375–381
Hull AW (1921) X-Ray crystal analysis of thirteen common metals. Phys Rev 17:571–588
Guillermet AF (1987) Critical evaluation of the thermodynamic properties of cobalt. Int J Thermophys 8:481–510
Kitakami O, Sato H, Shimada Y, Sato F, Tanaka M (1997) Size effect on the crystal phase of cobalt fine particles. Phys Rev B 56:13849
Braconnier L, Landrivon E, Clémençon I, Legens C, Diehl F, Schuurman Y (2013) How does activation affect the cobalt crystallographic structure? An in situ XRD and magnetic study. Catal Today 215:18–23
Ducreux O, Rebours B, Lynch J, Roy-Auberger M, Bazin D (2009) Microstructure of supported cobalt Fischer-Tropsch catalysts. Oil Gas Sci Technol Rev IFP 64:49–62
Srinivasan R, De Angelis RJ, Reucroft PJ, Dhere AG (1989) Structural characterization of cobalt catalysts on a silica support. J Catal 116:144–163
Li J-C (1996) Inelastic neutron scattering studies of hydrogen bonding in ices. J Chem Phys 105:6733–6755
Spencer E, Huang B, Parker SF, Kolesnikov A, Ross NL, Woodfield B (2013) The thermodynamic properties of hydrated γ-Al2O3 nanoparticles. J Chem Phys 139:
Parker SF, Refson K, Hannon AC, Barney E, Robertson SJ, Albers P (2010) Characterisation of hydrous palladium oxide: implications for low temperature carbon monoxide oxidation. J Phys Chem C 114:14164–14172
Petitto SC, Marsh EM, Carson GA, Langell MA (2008) Cobalt oxide surface chemistry: the interaction of CoO(100), Co3O4(110), and Co3O4(111) with oxygen and water. J Mol Cat A Chem 281:49–58
Tayal VP, Srivastava BK, Khandelwal DP, Bist HD (1980) Librational modes of crystal water in hydrated solids. Appl Spectrosc Rev 16:43–134
Parker SF, Bennington SM, Ramirez-Cuesta AJ, Auffermann G, Bronger W, Herman H, Williams KPJ (2003) Smith T Inelastic neutron scattering, Raman spectroscopy and periodic-DFT studies of Rb2PtH6 and Rb2PtD6. J Am Chem Soc 125:1165–11661
McInroy AR, Lundie DT, Winfield JM, Dudman CC, Jones P, Parker SF, Lennon D (2006) A temperature-programmed desorption, infrared and inelastic neutron scattering spectroscopic study of the interaction of hydrogen chloride with & #x03B7;-alumina. Catal Today 114:403–411
Albers PW, Weber W, Möbus K, Wieland SD, Parker SF (2016) Neutron scattering study of the terminating protons in the basic structural units of non-graphitising and graphitising carbons. Carbon 109:239–245
Parker SF, Leich V, Hönig J, Albers PW (2020) Investigation of commercial graphenes. ChemistryOpen 9:1060–1064
Larese JZ, Arnold T, Barbour A, Frazier LR (2009) Neutron investigations of rotational motions in monolayer and multilayer films at the interface of MgO and graphite surfaces. Langmuir 25:4078–4083
Mitchell PCH, Parker SF, Tomkinson J, Thompsett D (1998) Adsorbed states of dihydrogen on a carbon supported ruthenium catalyst: an inelastic neutron scattering study. J Chem Soc, Faraday Trans 94:1489–1493
Eckert J, Nicol JM, Howard J, Trouw FR (1996) Adsorption of hydrogen in Ca-exchanged Na-A zeolites probed by inelastic neutron scattering spectroscopy. J Phys Chem 100:10646–10651
Ramirez-Cuesta AJ, Mitchell PCH, Parker SF (2001) An inelastic neutron scattering study of the interaction of dihydrogen with the cobalt site of a cobalt aluminophosphate catalyst. Two-dimensional quantum rotation of adsorbed dihydrogen. J Mol Cat A Chem 167:217–224
Kubas GJ (2013) The art and beauty of inorganic synthesis on the path to discovery: transition metal coordination and activation of sulfur dioxide and dihydrogen. Comments Inorg Chem 33:102–121
Loewert M, Serrer M-A, Carambia T, Stehle M, Zimina A, Kalz KF, Lichtenberg H, Saraçi E, Pfeifer P, Grunwaldt J-D (2020) Bridging the gap between industry and synchrotron: an operando study at 30 bar over 300 h during Fischer-Tropsch synthesis. React Chem Eng 5:1071–1082
Rush JJ, Cavanagh RR, Kelley RD, Rowe JM (1985) Interaction of vibrating H-atoms on the surface of platinum particles by isotope-dilution neutron spectroscopy. J Chem Phys 83:5339–5341
Lehwald S, Rocca M, Ibach H, Rahman TS (1986) Surface phonon-dispersion of ordered overlayers. J Electron Spectrosc Relat Phenom 38:29–44
Erskine JL, Woods JP, Kulkarni AD, Dewette FW (1987) Surface vibrations on clean and hydrogen saturated W(100). J Elec Spec Rel Phen 44:2–36
Lennon D, Davidson AL, Parker SF (2017) The chemisorption of hydrogen over a high surface area cobalt catalyst: an INS investigation. STFC ISIS Neutron Muon Source. https://doi.org/10.5286/ISIS.E.RB1720059
Lennon D, Davidson AL, Parker SF, Webb P (2018) The chemisorption of hydrogen on cobalt. STFC ISIS Neutron Muon Source. https://doi.org/10.5286/ISIS.E.RB1820373
Lennon D, Parker SF, Webb P, Davidson AL (2019) The chemisorption of hydrogen over a Raney cobalt catalyst: an INS investigation. STFC ISIS Neutron Muon Source. https://doi.org/10.5286/ISIS.E.RB1920596
Albers PW, Parker SF (2019) Investigation of hydrogen/nickel and hydrogen/cobalt interactions in new metal foam and granulated Raney-type catalysts. STFC ISIS Neutron Muon Source. https://doi.org/10.5286/ISIS.E.RB1910579
Acknowledgements
The Science and Technology Facilities Council is thanked for the provision of neutron beam time.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Research Involved in Human or Animal Rights
There were no human or animal subjects involved in this research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davidson, A.L., Lennon, D., Webb, P.B. et al. The Characterisation of Hydrogen on Nickel and Cobalt Catalysts. Top Catal 64, 644–659 (2021). https://doi.org/10.1007/s11244-021-01425-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-021-01425-0