
Abstract
Residual biomass shows potential to be used as a feedstock for fast pyrolysis bio-oil production for energetic and chemical use. Although environmentally advantageous, further catalytic upgrading is required in order to increase the bio-oil stability, by reducing reactive compounds, functional oxygen-containing groups and water content. However, bio-oils may separate in fractions either spontaneously after ageing or by fractionated condensation. Therefore the effects of upgrading on different fast pyrolysis bio-oil (FPBO) fractions obtained from a commercially available FPBO were studied by elemental analysis, GC-MS and 1H-NMR. Not only the FPBO was upgraded by catalytic hydrotreatment, but also the heavy phase fraction formed after intentional aging and phase separation. The reactions were conducted between 175 and 325 °C and 80–100 bar by using a nickel–chromium catalyst in batch experiments. The influence of the hydrotreatment conditions correlated with the composition of the upgraded products. Higher oxygen removal was obtained at higher temperatures, whereas higher pressures resulted in higher hydrogen consumption with no significant influence on deoxygenation. At 325 °C and 80 bar 42% of the oxygen content was removed from the FPBO. Compounds attributed to pyrolysis oil instability, such as ketones and furfural were completely converted while the number of alcohols detected in the upgraded products increased. Coke formation was observed after all reactions, especially for the reaction with the fraction rich in lignin derivatives, likely formed by polymerization of phenolic compounds mainly concentrated in this phase. Independently of the feedstock used, the upgraded bio-oils were very similar in composition, with reduced oxygen and water content, higher energy density and higher carbon content.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The use of lignocellulosic biomass, especially residual biomass as a potential replacement for fossil resources has attracted the attention of the scientific community in the last years. The main interest lies in the production of fuels and/or chemicals, which is considered a second generation feedstock and thus avoids competition with food production [1]. A variety of lignocellulosic biomass can be valorized through thermochemical conversion for this purpose, such as wood, forest residues, wheat straw and other agriculture residues [2]. Among different biomass pyrolysis possibilities, by fast pyrolysis dry biomass is converted at temperatures of typically around 500 °C, short residence time in an inert atmosphere and ambient pressure, to result a brown viscous liquid, fast pyrolysis bio-oil (FPBO), as the main product along with some char and non-condensable gas [3, 4].
FPBO is a complex mixture of more than 300 mostly oxygenated compounds with different functional groups, such as carboxylic acids, aldehydes, furfurals, alcohols, carbohydrates, and ketones including high amounts of water. In addition, various lignin derivative compounds i.e. phenol and guaiacol are present, limiting its direct application as fossil fuel replacement [5,6,7]. The amount of these compounds is dependent on various parameters, in particular, the selected biomass as well as its moisture content. Reactor design, residence time, mixing behavior and heat transfer rates as well as condensation temperature also play a significant role in the product yield and composition, which can result in single-phase or multiphase pyrolysis condensates [8].
If separation in two phases occurs, a water rich phase consisting mostly of polar compounds such as organic acids, alcohols, aldehydes, ketones and sugar derivatives and a more dense organic phase with mostly high molecular compounds, such as phenolic, lignin-derived compounds are formed [8]. Also inorganic, heteroatom containing compounds may be present in the pyrolysis condensates.
Due to the complexity and the poor fuel quality of bio-oils, further treatment is required if renewable fuel and/or platform chemicals are intended to be produced. There are two principle ways: gasification of pyrolysis products to generate synthesis gas and subsequent production of synthetic fuels as e.g. targeted in KIT’s bioliq® process [9]. Alternatively, hydrotreatment has intensively been investigated as a promising method of bio-oil upgrading to directly produce drop-in fuels or fractions to be applied in crude oil refineries [10]. Using catalysts at elevated temperatures (150–450 °C) and hydrogen pressure (35–200 bar), the bio-oil can be upgraded by hydrodeoxygenation and hydrocracking to a product with lower viscosity, lower oxygen content as well as higher calorific value [11,12,13].
While the conversion of biomass into FPBO is well established and already commercialized, the upgrading still requires further investigation to overcome some of the unsolved challenges: the complex mixture of organic compounds associated with the presence of minerals or heteroatom containing organic molecules make the upgrading of pyrolysis-oils challenging. While minerals are attributed to catalyst poisoning [14, 15], the condensation of ketones, sugars and aldehydes at high temperatures are attributed to re-polymerization and formation of tar-like products, which can not only block the surface but also can result in plugging of continuous operation upgrading units [16].
For that reason, most of the effort dedicated so far is mainly focused on catalyst development. Early studies have considered the application of conventional catalysts such as CoMo/Al2O3 and sulphided NiMo/Al2O3 applied in hydrotreatment. Although very convenient, considering the current application of these catalysts in refineries [7], sulphided catalysts are rapidly deactivated by water and coke formation [6, 13] and may also introduce sulfur into the product.
Another category of catalysts that has been in the focus of many studies are heterogeneous non-noble metal catalysts. Investigations with transition metals, such as nickel, are of special interest due to its lower cost and reduced H2 consumption, compared with noble metals [17]. The combination of metals as bimetallic catalysts seems to be a good alternative for hydrodeoxygenation (HDO), considering its higher resistance to coke formation than monometallic catalysts [13]. Especially when combined in bimetallic catalysts, nickel shows interesting results: Boscagli et al. observed that a bimetallic nickel-copper catalyst (NiCu/Al2O3) showed better performance in bio-oil light phase HDO treatment compared to monometallic nickel catalysts on different supports, although poisoning by sulfur influenced the selectivity and activity of the catalyst [14]. For that reason, the choice of an appropriate catalyst for hydrotreatment is still a topic which demands efforts in order to find a catalyst with high activity, selectivity, sufficient stability and ability for regeneration, making the upgrading process feasible for industrial implementation [13].
The present work investigates the catalytic hydrotreatment of a FPBO and its fractions obtained by separation in two phases (light and heavy phase) after storage at elevated temperatures, considering that the long term storage or aging of bio-oils can result in natural separation in two phases. Therefore, the upgrading of the heavier fraction, rich in lignin derivative compounds was particularly investigated. Using a nickel chromium based catalyst we report the influence of temperature and pressure on the HDO activity.
2 Experimental
2.1 Feedstock: Fast Pyrolysis Bio-oil and Phase Rich in Lignin Derivatives
A intentionally aged (24 h, 80 °C) beech wood fast pyrolysis-oil spontaneously separated in two phases after storage was used in this study. The oil was composed by 59 wt% of light phase (LP) and 41 wt% of heavy phase (HP). Table 1 shows an overview of the main compounds in both fractions, obtained by solvent extraction [18]. While the LP concentrates most of the organic compounds rich in oxygen and water, the HP contains most of the lignin depolymerization products with lower oxygen content.
2.2 Catalyst
The catalyst selected for this study is a commercially available catalyst composed of Ni (30 wt%), NiO (26 wt%) and Cr2O3 (15 wt%), graphite (1.5 wt%) in diatomaceous earth support (27 wt%). A variety of analytical techniques was used in order to characterize the catalyst as described in Sects. 2.4 and 3.3.
2.3 Hydrotreatment Setup
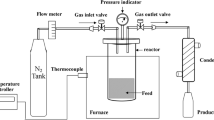
The reactions were performed in a 200 mL in-house designed and built autoclave for temperatures of up to 400 °C and pressure up to 360 bar. In order to promote a better transfer of hydrogen into the bio-oil, avoiding mass transfer limitation, the autoclave was equipped with a gas injection stirrer.
The experiments were performed at two different pressures (80 and 100 bar, set at room temperature), four different temperatures (175, 225, 275 and 325 °C) and two feedstocks: the original FPBO as well was as the heavy phase (HP). All the experiments were performed twice. Approximately 2.5 g of catalyst and 50 g of feedstock were added into the autoclave. For the experiments using FPBO, 20.5 g of HP and 29.5 g of LP were weighted (after phase separation, the phases were stored separately), according to the composition given in Sect. 2.1. The autoclave was closed, purged with nitrogen for 5 min for air removal and then pressurized with hydrogen. The autoclave was heated to the reaction temperature with a heating ramp of 5 °C/min. The reaction duration was 2 h. After the reaction, the autoclave was cooled down to approximately 40 °C using a pressurized air flow and then cooled down to approximately 20 °C using an ice bath. The gas sample was collected for further chemical analysis. The remaining mixture of upgraded liquid products, composed of upgraded light phase (ULP), upgraded oil phase (UO), solids and spent catalysts was collected, centrifuged (7000 rpm, 40 min, Thermo Scientific Heraeus Biofuge Stratos), separated and weighted. The spent catalyst was vacuum filtrated and washed with acetone several times, in order to remove any bio-oil residue.
The pressure was recorded before and after the reaction. The hydrogen consumed during the hydrotreatment was determined by using the ideal gas law, considering the amount of hydrogen before and after the reaction (determined by gas chromatography, Sect. 2.4) at room temperature [19, 20].
2.4 Characterization Techniques: Feedstock, Upgraded Products and Catalyst
The FPBO fractions (LP and HP) and upgraded products, upgraded light phase (ULP) and upgraded oil (UO) were characterized using the same analytical techniques. The elemental composition, hydrogen, carbon and nitrogen, was obtained by CHN 628 Leco. The oxygen was obtained by the difference. The high heating value (HHV) was obtained by Channiwala’s equation [21]:
Titration using the Karl Fischer Tritando 841, Metrohm was employed for water content determination; pH and density were obtained using a pH-meter 691, Metrohm and densimeter Anton Paar DMA4500M, respectively. Sulfur content and leached metals (upgraded products) were measured with an inductively coupled plasma optical emission spectrometer (ICP-OES, Agilent, 725).
Selected samples were analyzed using a gas chromatography–mass spectrometer (GC-MS HP G1800A) aiming a qualitative investigation of the main compounds in pre-selected samples. The GC-MS was equipped with a Restek stabilwax column (30 m × 0.25 mm × 0.25 µm) and the identification of the compounds was obtained matching the mass spectra with the NIST 2005 library. The sample was diluted 1:20 in methanol, filtrated (0.25 µm filter), injected (1 µL, split 1:20) at 250 °C (injector temperature) and measured using the temperature program starting at 40 °C (kept for 5 min), heated to 300 °C at 20 °C/min, (kept 20 min at this temperature) .
The distribution of functional groups was measured by proton nuclear magnetic resonance (1H-NMR). The samples were measured in a Bruker Biospin spectrometer composed by a 5.45 T magnet, 250 MHz frequency, pulse of 90°, 10.0633 s of acquisition time: 1.0 s of relaxation delay, 24 scans and 3255.2 Hz of spectral width and time domain of 32 k. The solutions were prepared diluting approximately 0.1 g of sample in deuterated methanol (about 800 µL) containing 2 g/L of internal standard (sodium 3-trimethylsilyl-2-2′,3,3′-tetradeuteropropionate). The spectra were integrated in pre-defined regions in order to calculate the millimoles of protons with respect to the internal standard. The software MestReNova was used for spectrum processing.
The gaseous products were quantified using a gas chromatograph Agilent 7890A equipped with two detectors—flame ionization detector (FID) and thermal conductivity detector (TCD)—and two columns—Restek Molsieve 5A and Restek 57096 Hayesep Q. The sample (100 µL, split 28:1) was injected at 250 °C (injector temperature) and measured using the temperature program starting at 50 °C kept for 10 min, heated to 90 °C at 3 °C/min, 20 °C/min to 150 °C, kept for 16 min, heated to 230 °C/min at 50 °C/min and kept at this temperature for 10 min.
The catalyst used in this study was characterized before and after the hydrotreatment reactions. Metals and sulfur content were obtained by inductively coupled plasma optical emission spectrometry (ICP-OES). The catalyst was digested in a microwave oven for 45 min in a mixture of 0.5 mL of hydrogen peroxide (H2O2), 2 mL of hydrochloric acid (HCl) and 6 mL of nitric acid (HNO3) prior the measurement. The X-ray diffraction spectra and the average crystallite size were obtained by the XRD (X’Pert PRO MPD PANalytical diffractometer, copper anode Cu Kα 1.54060 Å). The sample was measured in a 2θ range between 5° and 120° for 60 min and 0.017° step size. The crystalline size was obtained using the Scherrer equation (k factor = 0.9). The data were analyzed using the software X’PertHighscore Plus.
The specific surface area was determined by BET method (Belsorp Mini II), recording the nitrogen isotherm at 77 K. Fresh and spent catalysts were analyzed.
The carbon deposition over the spent catalysts was obtained by thermogravimetric analysis (Netzsch STA 409). The catalyst samples were heated in air (70 mL/min) from 20 to 105 °C at 10°C/min and kept for an hour (moisture determination); heated then to 1100 °C at 10 °C/min and finally kept at this temperature for 30 min [22]. The results of mass loss (discounting the mass loss attributed to moisture), were used to determine the amount of solids produced during the reactions.
The surface analysis of the fresh and spent catalysts was performed using a Scanning electron microscopy (SEM) (GeminiSEM 500, Zeiss; software: SmartSEM Version 6.01) with a thermal Schottky field emitter cathode. For the quantitative analysis of micro areas and the distribution of the elements, an energy dispersive X-ray spectrometer X-MaxN from Oxford with a silicon drift detector (80 mm2 and resolution of 127 eV) was employed (software: Aztec 3.3).
Furthermore the surfaces of the catalysts were analyzed by X-ray photoelectron spectroscopy (XPS). A layer of catalyst powder was uniformly distributed in a carbon tape and fixed in a molybdenum sample holder. The measurements were performed in an ultra-high vacuum chamber (base pressure 10−8 Pa) equipped with an unmonochromated XR-50 Mg K alpha X-ray source and a Phoibos 150 analyzer (manufacturer SPECS). The angle between the analyzer and the X-ray source was 45°. The electrons originating from a samples were detected along the surface normal of the sample (sample area: 2 mm diameter); the energy scale was calibrated using the Ag 3d peak of a silver reference sample. Peak shifts due to charging effects were compensated using the flood gun FG 15/40 (SPECS). The chemical composition was quantified with the software CasaXPS [23], using the integrated database for sensitivity factors. For the quantification, a Shirley background was subtracted from the peaks Ni 2p 3/2, Cr 2p, and C1s. For O 1s, Si 2s, and Al 2s a linear background was assumed.
3 Results and Discussion
3.1 Hydrotreated Products Distribution
The products distribution for each condition tested is summarized in Table 2. Mainly two product phases are obtained, i.e. the upgraded light phase (ULP) and the upgraded oil phase (UO). Gas and solid phase were present in lower concentrations. Comparing firstly the reaction performed at 175, 225 and 275 °C with FPBO, the oil yield slightly increased with the increase of temperature for reactions performed at 80 bars whereas a small reduction in the oil yield was observed at 100 bars. In general, for these conditions the oil yield amounted to 39.3–43.7 wt% with higher UO yields at 275 °C and 80 bar (43.7 wt%) and lower water concentration (Table 3).
The products obtained using the isolated heavy phase (HP) showed the opposite behavior: the higher the temperature, the lower the UO yield. Among the reactions performed at 175, 225 and 275 °C, the lower yields of UO were observed for the reactions performed at 275 °C and 80 bar (65.5 wt%) resulting also in an UO with the lower water concentration (7.2 wt%) compared to the reactions performed at 275 °C, 100 bar (7.5 wt%).
The following reaction performed at 325 °C, 80 bar for FPBO feedstocks showed UO yields (43.6 wt%) comparable to the results with 275 °C, 80 bar (43.7 wt%). At the same time, the upgrading at this condition resulted in an UO with the lowest water content observed for this feedstock (6.5 wt%). The upgrading of the HP at this conditions also resulted in an UO with the lowest water concentration for this feedstock and the lowest UO yield (63.10 wt%), which can also be attributed to the lowest water content.
Regarding the gas production, an increase in the gas yield was observed with the temperature. For reactions performed at 325 °C and 80 bar, the gas production was approximately ten times higher than the gas yield at 175 °C, for both feedstocks. The gas composition is further discussed in the Sect. 3.2.
For all of the reactions performed, the solid concentration ranged between 0.44 and 1.28 wt%. Although a small amount of solid products was observed, the higher yields were obtained for the reactions using the isolated HP. This can be related to the fact that higher concentration of oligomeric phenolic compounds in the HP might lead to a higher amount of polymerization products, resulting in higher amount of coke formation [24,25,26]. In general the losses obtained during the reaction were below 12 wt% and are mainly due to upgraded products which could not be completely recovered from the reactor walls.
3.2 Upgraded Products
The upgraded liquid products were analyzed in detail. The elemental composition of the products (dry basis) obtained for both feedstocks are summarized in Tables 3 and 4. The concentration of carbon in the UOs increased according to the upgrading temperature whereas the oxygen content decreased.
Firstly, the hydrotreatment reactions of both feedstocks were evaluated at 175, 225 and 275 °C at 80 and 100 bar. At these conditions the carbon was mainly concentrated in the UO for both feedstocks (63.13–68.69 wt%), resulting in an increase in the energy density in the upgraded products (28.06–32.37 MJ/Kg) in comparison to the feedstocks (FPBO = 24.33 MJ/Kg and HP = 27.23 MJ/Kg). The hydrogen consumption varied from 205 NL/Kg of feed at 175 °C at 80 bar to 365 NL/Kg of feed at 275 °C, 100 bar.
Although a higher hydrogen consumption was observed for the reactions performed at 100 bars in comparison to the reactions performed at 80 bars for the same temperature, no significant effect of pressure on the hydrodeoxygenation was observed for both feedstocks. For example, the degree of deoxygenation (DOD) obtained at 275 °C, 80 bars for the upgrading of FPBO was 40.7% while at 100 bar the DOD obtained was 38.0%. Consequently, we consider that the temperature exhibits a higher influence on the hydrodeoxygenation degree.
Hence, additional reactions were performed at 325 °C and 80 bars. At this condition the carbon content in the UOs increased (UOFPBO, 325 °C, 80 bar = 70.4 wt%; UOHP, 325 °C, 80 bar = 71.6 wt%) in comparison to the other conditions tested. Furthermore the DOD was also higher compared to the reactions performed previously. For the FPBO a reduction of 42.05% in the oxygen content was observed while for the HP the DOD achieved was 32.0% (Table 4).
Comparing the results of the experiments at 325 °C, 80 bar and 275 °C, 100 bars confirms that a temperature increase of 50 °C has more beneficial effects than a pressure increase of 20 bars: At 325 °C, 80 bar DOD, the H content of UO and the H/C ratio of UO is higher, whereas 22–28% less H2 is consumed compared to the reaction at 275 °C, 100 bar.
The pH of the upgraded products remains very similar in the feeds while the density decreased in comparison to the feedstocks (pHHP = 3.1 ρHP = 1.19 g/cm3; pHLP = 2.8 ρLP = 1.18 g/cm3). For the upgrading at 325 °C, 80 bar FPBO the upgraded oil showed a pHUO = 3.2 and ρUO = 1.08 g/cm3 and the ULP showed pHULP = 3.2 ρULP = 1.02 g/cm3. The upgraded products of the reaction with HP showed pHUO = 3.7 ρUO = 1.09 g/cm3 and pHULP = 3.4 ρULP = 1.02 g/cm3 respectively.
For all evaluated conditions, the water content in the UOs dropped with higher temperature, while it increased in the light phases considerably (41.5–80.1 wt%). Furthermore, for all the upgraded products, including ULP and UO, the nitrogen content was below 1 wt% and therefore no significant changes could be detected.
The changes in the distribution of functional groups as a function of the upgrading reaction conditions were determined by 1H-NMR. For comparison purposes, the ULP will be related to the initial LP whereas the UO will be compared to the initial HP. The same tendencies were observed for the reactions performed at 80 and 100 bar; hence, the results obtained at 100 bar are only reported in the Supplementary Material (Figure S.5).
The integration regions for the upgraded oils (Fig. 1a, b) show that the concentration of protons belonging to the alkane region (0.5–1.5 ppm) increased significantly in the UOs (16.78–22.68 mmol/g sample) compared to the initial concentration of the HP (8.25 mmol/g sample), as well as the proton concentration in the region of α proton to carboxylic acid or keto-group, α proton to unsaturated group (feedHP = 17.81 mmol/g sample; products = 18.23 to 32.54 mmol/g sample).

1H-NMR spectra integration of the upgraded products at different temperatures and 80 bar. a UO, feed: FPBO; b UO, feed: HP; c ULP, feed: FPBO; d ULP, feed: HP
The concentration of protons in the alcohols, ethers and alkenes (3.0–4.3 ppm) regions increase with the temperature up to 225 °C. For the reactions performed at higher temperatures (275 and 325 °C) the concentration of protons then decreased (11.76–8.63 mmol/g sample) for values below the feed (HP = 13.17 mmol/g sample). The decrease in the concentration of protons at higher temperatures can be attributed to the fact that compounds such as alcohols, ethers and alkenes tend to react at approximately 250–300 °C, as also reviewed by Elliot [27].
The concentration of protons belonging to the carbohydrates, water and O–H exchanging groups (4.3–6.0 ppm) showed a considerable reduction compared to the feed (HP). A clear tendency is observed with the increase of the temperature: the higher the temperature, the lower the concentration of protons in this region. Such a tendency can be explained considering that at higher temperatures the water content in the upgraded oil is reduced; in addition, conversion of sugars molecules during the hydrotreatment could also contribute to the reduction of protons in this region [17, 28]. A reduction of protons in the region of (hetero)-aromatics was observed for all upgraded oils in comparison to the feed, although the concentration in the UO was similar for all the conditions tested. The only exception was observed to the oil upgraded at 325 °C, 80 bar, feed: HP, which showed the higher concentration of protons in this region (8.29 mmol/g sample), with a slightly higher concentration. Protons attributed to aldehydes (9.5–10.1 ppm) were present in very small amount in the feed and were not observed in the products; these reactive compounds usually react at mild temperatures and can be reduced completely by hydrogen [27, 29].
The integration regions for the upgraded light phases (Fig. 1c, d) show that the concentration of protons belonging to the alkane region (0.5–1.5 ppm) increased in the ULPs in comparison to the LP (0.056 mmol/g sample), especially in reactions performed at lower temperatures. At 325 °C, the concentration of protons is the lowest compared to all the conditions evaluated, which could indicate migration of these compounds to the upgraded oil. The protons concentration in the region between 1.5 and 3.0 ppm has also increased in comparison to the original LP (3.26 mmol/g sample).
The concentration of protons in the range of 3.0–4.3 (alcohols, ethers and alkenes) reduced significantly in comparison to the feed (LP = 50.25 mmol/g sample), especially for the reactions performed at 325 °C, 80 bar (FPBO = 8.26 mmol/g ULP). It can be assumed that there is a tendency of alcohols, ether and alkenes of being converted at higher temperatures.
The protons in the range of 4.3–6.0 ppm increased considerably in the ULPs in comparison to the LP of the feed (13.88 mmol/g sample). Furthermore the highest concentrations were observed for the reactions performed at more severe conditions (325 °C, 80 bars). This could be explained considering that the water removed from the upgraded product is concentrated in the ULPs; the higher the temperature the lower the water concentration in the upgraded oil and consequently the higher the water concentration in the upgraded light phase. For example, this assumption is in agreement with the results observed to the reactions applying the heavy phase at 325 °C and 80 bar: the highest water concentration was observed in the ULPs at this condition (80.1 wt%), in agreement with the highest concentration of protons (95.91 mmol/g ULP) among all the ULPs evaluated.
The protons in the (hetero)-aromatic range also decreased considerably in the upgraded products while aldehydes, initially present in the LP, were not observed in the ULPs.
In order to identify the main compounds obtained at different reaction conditions, as well as to compare the feedstock and upgraded products, a qualitative investigation by GC–MS was performed. The main compounds in selected samples are depicted in Fig. 2 and correlated with the retention time given in the Table 5. More detailed information, including the chromatograms for all the conditions tested is available in the Supplementary Material (Figures S.1, S.2, S.3 and S.4).

Chromatograms obtained for upgraded oils (a) and upgraded light phases (b) in comparison to the feeds (LP and HP)
Some relevant peaks were identified and correlated with the data given in Table 5. Furthermore the main reaction pathways identified are available in the Figure S.13. The appearance of alcohols in cyclic structure in the UOs as well as in the ULPs, such as cyclopentanol, 2-methyl-cyclopentanol, and cyclohexanol is observed in the upgraded product while ketones (1-hydroxy-2-propanone; 2-cyclopenten-1-one; 2-methyl-2-cyclopenten-1-one), initially present in the feeds, are not observed in any of the upgraded products. This indicates that keto groups reacted mainly by hydrogenation of the carbonyl groups producing alcohols [7]. Furfural was converted for all of the reaction conditions tested. The presence of tetrahydrofurfuryl alcohol (RT: 16.14 min) in the UOs and ULPs, observed for the reactions performed at T > 175 °C, indicates that furfural was hydrogenated to tetrahydrofurfuryl alcohol [7, 30]. This selectivity was previously observed by Boscagli et al. [17] for a Ru/C catalyst while for a bimetallic nickel catalyst (NiCu/Al2O3) the ring opening seems to be more favorable. Further investigations are presently undertaken to investigate whether this significant difference in selectivity may be the results of chromium in the catalyst.
Propylene glycol was present in the upgraded products, especially in the ULPs. Most likely hydroxypropanone (RT: 12.46 min), detected in the feeds but not in the upgraded products, was hydrogenated to propylene glycol. Furthermore, the increase in the propylene glycol, and other diols such as 1,2-butanediol (Figures S.1 and S.2) can be associated to the sugar molecules of the feedstock; the hydrogenation of xylose and glucose leads to the formation of xylitol and sorbitol which can undergo further hydrogenolysis resulting in the alcohols such as propylene glycol, 1,2-ethanediol and 1,2-butanediol [28], present in the ULPs.
Aromatic compounds such as 2-methoxyphenol (guaiacol) and 2-methoxy-4-ethylphenol (4-ethylguaiacol) were identified in the products as well as in the feed. Molecules with higher retention time, such as eugenol, vanillin and isoeugenol were not identified in the products. Vanillin, derived of the guayacyl units in the polymeric lignin structure [31] and observed in the feed, was completely converted after the reactions. It can be converted to vanyllil alcohol by hydrogenation and then to guaiacol by demethoxylation [32, 33]. Guaiacol can further undergo demethoxylation and hydrogenation resulting in cyclic alcohols [29, 34]. For the whole temperature range, eugenol was completely converted: the hydrogenation of the double bonds leads to the conversion to propylguaiacol, identified in the upgraded products (RT: 25.22 min) [29]. A small peak related to phenol was observed in the chromatogram of the HP. After the reaction the peak disappeared, suggesting that phenol was completely converted: the hydrogenation of the aromatic ring of phenol leads to the conversion to cyclohexanol (compound detected in the upgraded products) [17, 27]. Moreover the peak at 25.22 min in the upgraded products can also suggest the hydrogenation of the double bond in the isoeugenol molecule.
For the conditions tested, the hydrogenation of the molecules was more likely to occur compared to hydrodeoxygenation. Furthermore, the compounds identified in the upgraded light and heavy phases were for all the reaction conditions very similar. Keto groups, furfural and compounds such as vanillin and eugenol were completely converted.
The main compounds in the gas phase (excluding hydrogen, which was the main compound detected), are presented in Table 6. CO2 was the main compound, followed by methane, and in smaller amounts ethane, propane and CO.
The concentration of CO2 increased with the temperature for both feedstocks, with a slightly higher concentration for the reactions with (FPBO). The reactions performed at higher pressures lead to lower amounts of CO2, when comparing reactions performed at the same temperature. CO2 is mainly obtained by decarboxylation, whereas CO forms by decarbonylation [10]. CH4 can be produced as a result of carbohydrates conversion, C–C bonds cleavage as well as methanation of CO2 [7, 17].
3.3 Catalyst Characterization
Based on the results above, the spent catalyst of reactions conducted with FPBO and HP at 325 °C and 80 bar was selected for further characterization, considering the higher degree of deoxygenation, chemical composition and hydrogen consumption (reducing excess of hydrogen consumption), respectively. The characterization of fresh catalyst is presented for comparison.
The metal leaching was obtained by ICP-OES measuring the content of Ni and Cr in the ULPs. 0.73 wt% of Ni (in relation to the initial concentration of metal in the catalyst) was leached after the reaction with FPBO whereas 0.43 wt% was leached for the reaction with HP. The high amount leached for the FPBO can be related to the higher water concentration in this feedstock [14, 35]. The amount of chromium leached was lower compared to Ni; 0.010 wt% of Cr was leached for FPBO feed and 0.014 wt% for reaction with HP.
The amount of solid deposition over the catalyst was measured by TGA and the results were presented in Sect. 3.1, as the percentage of solid formed. The TGA measurements area available in the Supplementary Material (Figure S.14). The catalyst used for HP upgrading showed after the reaction approximately the double of the solid deposition compared to the catalyst used for the FPBO upgrading and also smaller specific surface area (Table 7) although approximately 80% of the original specific surface area was preserved. Furthermore, in order to investigate the surface of the catalysts after the reaction, the approximate composition of the fresh and spent catalysts was analyzed by SEM-EDX.
The elemental distribution of the catalyst is shown in Table 7. The carbon content in the spent catalyst increased in relation to the fresh catalyst, due to coke deposition, especially for the catalyst used in the HP upgrading, in agreement with the solid residue results (Table 7). Condensation of phenolic compounds (present in higher concentration in the heavy phase), can lead to ring condensation, being responsible for the coke formation [36, 37]. The higher concentration of these compounds in the heavy phase (Table 1) could result in higher amounts of coke, as observed. Furfural, present is both feedstocks, tends to polymerize under high temperatures, which could also contribute to formation of the solid deposition observed [22, 38].
Furthermore, catalyst poisons were detected in the spent catalysts. Sulfur was present in the catalysts tested in both conditions, in very similar concentration, despite of their different concentration in the feedstocks (0.012 wt% FPBO; 0.009 wt% HP). Due to the poisoning effect over Ni catalysts the presence of sulfur should be taken into account especially if long term applications are intended [14]. This topic will be object of future investigation.
Compounds such as Mg and Ca were also observed in the spent catalyst. Magnesium was present in higher concentration for the reaction with FPBO whereas higher concentration of calcium was observed for the spent catalyst applying HP. Usually compounds such as magnesium and calcium are present in small concentrations in pyrolysis-oils and are attributed to catalyst poisoning, reducing its activity in the same way that sulfur and nitrogen compounds [14].
The X-ray powder diffraction patterns are depicted in Fig. 3. The passivated/fresh catalyst shows broad reflections which could indicate a highly dispersed metal distribution [39]. Reflections attributed to metallic nickel could be observed in the passivated/fresh catalyst (2θ = 44.5°, 51.8°, 76.4°, 92.95° and 98.45°) as well as small broad reflections attributed to nickel oxide (2θ = 37.2°, 42.6° and 62.8°). Although present in high concentration (Cr: 12.8 wt%), reflections attributed to Cr2O3 (JCPDS 381479) could not be observed, indicating a high dispersion of chromium oxide or an amorphous chromium phase [17, 39, 40]. After the reaction, the catalysts show sharper reflections attributed to metallic nickel for both conditions tested, indicating the reduction of the nickel oxide under the reductive conditions of the upgrading reactions.

X-ray powder diffraction patterns of fresh and spent catalysts
The average crystallite size estimated by Scherrer equation increased after the reaction. While the average crystallite size for the fresh catalyst was about 4.4 nm, the spent catalysts used for HP and FPBO upgrading increased to 18 and 16 nm, respectively. This indicates some sintering of the Ni particles [14], as previously observed for nickel catalyst at high temperature [41].
Further surface characterization of the catalyst was performed by XPS. Figure 4a shows the survey spectra of the fresh and the spent catalysts. The characteristic XPS peaks for Ni, Cr, O, and Si are indicated. For all samples, the Cr 2p3/2 peak was observed at 276 eV, as expected for Cr2O3 [39, 42, 43]. Figure 4b shows a zoom into the Ni 2p region. The Ni 2p3/2 peak was observed at 855 eV, with a pronounced satellite peak at 861 eV. [44], reported similar peak positions for Ni(OH)2 and NiO. The small peak around 846 eV, visible only for the intense peak of the fresh catalyst, originates from the Mg Kα3 line of the non-monochromatized X-ray beam. For the spent catalyst, an additional shoulder was observed at 851.5 eV (indicated by an arrow), which is close expected peak position of metallic nickel at 852.6 eV [44, 45]. This is consistent with XRD observations suggesting a reduction of NiO to Ni for the spent catalysts.

a XPS spectra for the fresh and the spent catalysts. b Zoom into the Ni2p region (offset adapted for better comparison)
The O 1s peak at 530.5 eV results from a superposition of NiO, Cr2O3, silica (support material for the catalyst), and adventitious oxygen due to the sample exposure to air [44, 46].The C 1s peak at 284 eV is also a superposition of carbonaceous deposition due to the catalytic process, adventitious carbon. However, in agreement with the EDX measurements the carbon content determined by XPS is significantly increased for the spent catalysts, indicating that for the spent catalysts the C 1s peak is dominated by the carbonaceous deposition. Due to the lower concentration of species previously observed by SEM-EDX and the limitation of the XPS to concentrations below 0.1 atomic percentage (at.%), S, Ca and Mg were not identified in the XPS of spent catalyst samples.
The chemical composition within the top most few nm of the catalyst samples was determined assuming a homogeneous material distribution within this layer. The relative atomic concentration is summarized in Table 8. In agreement with the EDX results, the carbon content increased for the spent catalysts. The reduction of the oxygen content is consistent with the reduction of nickel oxide species to metallic nickel, as observed by XRD. However, in contrast to the EDX results and the low degree of metal leaching, the Cr/Ni ratio increased significantly for the spent catalyst. This can be explained with the larger Ni grain size observed for the spent catalyst or encapsulation of Ni by carbonaceous compounds. With increasing grain size, the surface/volume ratio decreases. XPS is only sensitive to the surface region, underestimating thus the Ni content of the spent catalyst. In addition to the main components of the catalyst, small amounts of silicon and aluminum where detected. They can be are attributed to the support material.
4 Conclusion
The complete beech wood pyrolysis-oil and the isolated heavy phase were upgraded at different conditions of temperature and pressure applying a nickel chromium catalyst. The hydrotreatment of both feedstocks resulted in upgraded oils with higher carbon content and lower oxygen content, mainly at higher temperatures (275–325 °C). At higher pressure (100 bar compared to 80 bar), higher consumption of hydrogen was observed with no significant reduction in the oxygen content, indicating mainly hydrogenation reactions are favored thereby not hydrodeoxygenation. Higher gas production was observed for the reactions at higher temperatures, resulting in a gas product composed by carbon dioxide (decarboxylation product) and methane (C–C bond cleavage) for both feedstocks. The 1H-NMR showed an increase in the aliphatic region by hydrogen treatment whereas the aromatics remained in the upgraded oil. Furthermore, gas chromatography showed that compounds such as ketones, furfural and aldehydes were completely converted while aromatics were stable, which is in agreement with the 1H-NMR results. The increase in the number of alcohols detected can indicate hydrogenation of the carbonyl groups.
The higher solid deposition over the spent catalyst was observed for the catalyst used for HP upgrading although no significant reduction in the specific surface area was observed. It is assumed that the higher concentration of phenolic compounds in the HP leads to condensation reactions of the aromatic rings resulting in coke formation. In addition to carbon, small amounts of poisoning substances such as sulphur, calcium and magnesium were also detected in the spent catalysts. Furthermore, small amounts of leached metal were observed in the upgraded light phase.
In summary, the results demonstrate that the upgrading of complete pyrolysis oil or isolated heavy phase leads to upgraded oils with very similar properties, with lower oxygen and water content and higher energy density. The nickel chromium catalyst was active for the conditions tested and further investigations will elucidate the influence of poisoning substances, such as sulfur, carbon and magnesium over the long term stability of the catalyst.
References
Alonso DM, Wettstein SG, Dumesic JA (2012) Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem Soc Rev 41:8075–8098. https://doi.org/10.1039/C2CS35188A
Laurichesse S, Avérous L (2014) Chemical modification of lignins: towards biobased polymers. Prog Polym Sci 39:1266–1290. https://doi.org/10.1016/j.progpolymsci.2013.11.004
Bridgwater AV (2012) Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 38:68–94. https://doi.org/10.1016/j.biombioe.2011.01.048
Al Arni S (2018) Comparison of slow and fast pyrolysis for converting biomass into fuel. Renew Energy 124:197–201. https://doi.org/10.1016/j.renene.2017.04.060
Bridgwater AV (2013) Report with a process scheme for hydrotreating catalytically produced bio-oil and esterifying bio-oil with alcohols and producing fuel gas for power production or synfuels production. pp 1–54
Gunawan R, Li X, Lievens C et al (2013) Upgrading of bio-oil into advanced biofuels and chemicals. Part I. Transformation of GC-detectable light species during the hydrotreatment of bio-oil using Pd/C catalyst. Fuel 111:709–717. https://doi.org/10.1016/j.fuel.2013.04.002
He Z, Wang X (2012) Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal Sustain Energy 1:28–52. https://doi.org/10.2478/cse-2012-0004
Oasmaa A, Fonts I, Pelaez-Samaniego MR et al (2016) Pyrolysis oil multiphase behavior and phase stability: a review. Energy Fuels 30:6179–6200. https://doi.org/10.1021/acs.energyfuels.6b01287
Funke A, Richter D, Niebel A et al (2016) Fast pyrolysis of biomass residues in a twin-screw mixing reactor. J Vis Exp. https://doi.org/10.3791/54395
Mortensen PM, Grunwaldt JD, Jensen PA et al (2011) A review of catalytic upgrading of bio-oil to engine fuels. Appl Catal A 407:1–19. https://doi.org/10.1016/j.apcata.2011.08.046
Venderbosch R, Heeres H (2011) Pyrolysis oil stabilisation by catalytic hydrotreatment. In: dos Santos Bernardes MA (ed) Biofuel’s engineering process technology. InTech, Rijeka, pp 385–410
Al-muhtaseb AH, Jamil F, Baawain M et al (2016) Bio-oil upgrading by catalytic cracking over different solid catalysts. J Adv Catal Sci Technol 3:12–19
Saidi M, Samimi F, Karimipourfard D et al (2014) Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ Sci 7:103–129. https://doi.org/10.1039/C3EE43081B
Boscagli C, Yang C, Welle A et al (2017) Effect of pyrolysis oil components on the activity and selectivity of nickel-based catalysts during hydrotreatment. Appl Catal A Gen 544:161–172. https://doi.org/10.1016/j.apcata.2017.07.025
Mortensen PM, Gardini D, Carvalho HWP de et al (2014) Stability and resistance of nickel catalysts for hydrodeoxygenation: carbon deposition and effects of sulfur, potassium, and chlorine in the feed. Catal Sci Technol. https://doi.org/10.1039/c4cy00522h
Wang H, Wang Y (2016) Characterization of deactivated bio-oil hydrotreating catalysts. Top Catal 59:65–72. https://doi.org/10.1007/s11244-015-0506-6
Boscagli C, Raffelt K, Zevaco TA et al (2015) Mild hydrotreatment of the light fraction of fast-pyrolysis oil produced from straw over nickel-based catalysts. Biomass Bioenergy 83:525–538. https://doi.org/10.1016/j.biombioe.2015.11.003
Schmitt CC, Boscagli C, Rapp M, Raffelt R, Dahmen N (2017) Characterization of light and heavy phase of pyrolysis-oil from distinct biomass for further upgrading reactions. In: Proceedings of European Biomass Conference and Exhibition. https://doi.org/10.5071/25thEUBCE2017-3AV.3.15
Chen W, McClelland DJ, Azarpira A et al (2016) Low temperature hydrogenation of pyrolytic lignin over Ru/TiO 2: 2D HSQC and 13 C NMR study of reactants and products. Green Chem 18:271–281. https://doi.org/10.1039/C5GC02286J
Ardiyanti AR, Khromova SA, Venderbosch RH et al (2012) Catalytic hydrotreatment of fast pyrolysis oil using bimetallic Ni-Cu catalysts on various supports. Appl Catal A Gen 449:121–130. https://doi.org/10.1016/j.apcata.2012.09.016
Channiwala SA, Parikh PP (2002) A unified correlation for estimating HHV of solid, liquid and gaseous fuels. Fuel 81:1051–1063. https://doi.org/10.1016/S0016-2361(01)00131-4
Reddy Kannapu HP, Mullen CA, Elkasabi Y, Boateng AA (2015) Catalytic transfer hydrogenation for stabilization of bio-oil oxygenates: reduction of p-cresol and furfural over bimetallic Ni-Cu catalysts using isopropanol. Fuel Process Technol 137:220–228. https://doi.org/10.1016/j.fuproc.2015.04.023
Fairley N, Carrick A (2005) The Casa Cookbook: recipes for XPS data processing, 1st edn. Acolyte Science, Knutsford
Joshi N, Lawal A (2012) Hydrodeoxygenation of pyrolysis oil in a microreactor. Chem Eng Sci 74:1–8. https://doi.org/10.1016/j.ces.2012.01.052
Boscagli C, Raffelt K, Grunwaldt JD (2017) Reactivity of platform molecules in pyrolysis oil and in water during hydrotreatment over nickel and ruthenium catalysts. Biomass Bioenergy 106:63–73. https://doi.org/10.1016/j.biombioe.2017.08.013
Li X, Gunawan R, Wang Y et al (2014) Upgrading of bio-oil into advanced biofuels and chemicals. Part III. Changes in aromatic structure and coke forming propensity during the catalytic hydrotreatment of a fast pyrolysis bio-oil with Pd/C catalyst. Fuel 116:642–649. https://doi.org/10.1016/j.fuel.2013.08.046
Elliott DC (2007) Historical developments in hydroprocessing bio-oils. Energy Fuels 21:1792–1815. https://doi.org/10.1021/ef070044u
Gandarias I, Arias PL (2013) Hydrotreating catalytic processes for oxygen removal in the upgrading of bio-oils and bio-chemicals. Liq Gaseous Solid Biofuels-Convers Tech. https://doi.org/10.5772/50479
Jongerius AL (2013) Catalytic conversion of lignin for the production of aromatics
Resasco DE, Sitthisa S, Faria J et al (2011) Furfurals as chemical platform for biofuels production. Solid Waste Renew Resour
Holladay JE, White JF, Bozell JJ, Johnson D (2007) Top value-added chemicals from biomass volume II: results of screening for potential candidates from biorefinery lignin. Pac Northwest Natl Lab II. https://doi.org/10.2172/921839
Yang X, Liang Y, Cheng Y et al (2014) Hydrodeoxygenation of vanillin over carbon nanotube-supported Ru catalysts assembled at the interfaces of emulsion droplets. Catal Commun 47:28–31. https://doi.org/10.1016/j.catcom.2013.12.027
Kayalvizhi J, Pandurangan A (2017) Hydrodeoxygenation of vanillin using palladium on mesoporous KIT-6 in vapour phase reactor. Mol Catal 436:67–77. https://doi.org/10.1016/j.mcat.2017.04.002
Wang X (2013) Valorization of lignin and bio-oil by catalytic hydrogenation with Ni catalyst. Fakultät für Chemie der Ruhr Universität Bochum
Huynh TM, Armbruster U, Nguyen LH, Nguyen DA (2015) Hydrodeoxygenation of bio-oil on bimetallic catalysts: from model compound to real feed. J Sustain Bioenergy Syst 5:151–160. https://doi.org/10.4236/jsbs.2015.54014
Gholizadeh M, Gunawan R, Hu X et al (2016) Effects of temperature on the hydrotreatment behaviour of pyrolysis bio-oil and coke formation in a continuous hydrotreatment reactor. Fuel Process Technol 148:175–183. https://doi.org/10.1016/j.fuproc.2016.03.002
Valle B, Aramburu B, Santiviago C et al (2015) Upgrading of bio-oil in a continuous process with dolomite catalyst. Energy Fuels 28:2014–2015. https://doi.org/10.1021/ef501600f
Nakagawa Y, Tamura M, Tomishige K (2015) Catalytic conversions of furfural to pentanediols. Catal Surv Asia 19:249–256. https://doi.org/10.1007/s10563-015-9194-2
Gómez-Cazalilla M, Infantes-Molina A, Mérida-Robles J et al (2009) Chromium species as captors of sulfur molecules on nickel-based hydrotreating catalysts. Energy Fuels 23:101–110. https://doi.org/10.1021/ef800741n
Marchetti L, Miserque F, Perrin S, Pijolat M (2015) XPS study of Ni-base alloys oxide films formed in primary conditions of pressurized water reactor. Surf Interface Anal 47:632–642. https://doi.org/10.1002/sia.5757
Olarte MV (2011) Base-catalyzed depolymerization of lignin and hydrodeoxygenation of lignin model compounds for alternative fuel production
Biesinger MC, Brown C, Mycroft JR et al (2004) X-ray photoelectron spectroscopy studies of chromium compounds. Surf Interface Anal 36:1550–1563. https://doi.org/10.1002/sia.1983
Deutsch KL (2012) Copper catalysts in the C-O hydrogenolysis of biorenewable compounds. PhD Thesis
Biesinger MC, Payne BP, Lau LWM et al (2009) X-ray photoelectron spectroscopic chemical state Quantification of mixed nickel metal, oxide and hydroxide systems. Surf Interface Anal 41:324–332. https://doi.org/10.1002/sia.3026
Prieto P, Nistor V, Nouneh K et al (2012) XPS study of silver, nickel and bimetallic silver-nickel nanoparticles prepared by seed-mediated growth. Appl Surf Sci 258:8807–8813. https://doi.org/10.1016/j.apsusc.2012.05.095
Payne BP, Biesinger MC, McIntyre NS (2012) Use of oxygen/nickel ratios in the XPS characterisation of oxide phases on nickel metal and nickel alloy surfaces. J Electron Spectrosc Relat Phenom 185:159–166. https://doi.org/10.1016/j.elspec.2012.06.008
Acknowledgements
The authors are grateful to the Bioeconomy Graduate Program - BBWForwerts, Brazilian National Council for Science and Technology (CNPQ) and BeMundus for the financial support. The authors are also grateful to Michael Zimmermann, Herman Köhler, Pia Griesheimer, Gilbert Zwick, Petra Janke and Jessica Heinrich for the support with analytical techniques.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schmitt, C.C., Raffelt, K., Zimina, A. et al. Hydrotreatment of Fast Pyrolysis Bio-oil Fractions Over Nickel-Based Catalyst. Top Catal 61, 1769–1782 (2018). https://doi.org/10.1007/s11244-018-1009-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-018-1009-z