
Abstract
Pore condensation has been suggested as an initiator of deactivation in the dehydration of glycerol to acrolein. To avoid potential pore condensation of the glycerol, a series of WO3 supported on ZrO2 catalysts have been prepared through thermal sintering, with modified pore systems. It was shown that catalysts heat treated at temperatures above 800 °C yielded suitable pore system and the catalyst also showed a substantial increase in acrolein yield. The longevity of the heat-treated catalysts was also improved, indeed a catalyst heat treated at 850 °C displayed significantly higher yields and lower pressure-drop build up over the 600 h of testing. Further, the catalyst characterisation work gave evidence for a transition from monoclinic to triclinic tungsten oxide between 850 and 900 °C. There is also an increase in acid-site concentration of the heat-treated catalysts. Given the improved catalyst performance after heat-treatment, it is not unlikely that pore condensation is a significant contributing factor in catalyst deactivation for WO3 supported on ZrO2 catalysts in the glycerol dehydration reaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In the wake of the biodiesel boom, there is a large interest in the side-product glycerol as a starting point for fuel and chemical synthesis [1, 2]. The lowest cost quality of glycerol is the so-called crude grade, which is obtained via esterification of triglycerides with methanol, resulting in fatty acid methyl esters and glycerol. The crude grade will contain not only the glycerol, but also the sodium hydroxide used as a catalyst, other salts, some water as well as methanol, mono-, di-, and triglycerides and matter organic non-glycerol (MONG) in different proportions depending on source [2]. In synthesising chemicals and fuels, there is a desire to keep cost down, and at the same time meet product specifications. There is quite some clean-up required before using the glycerol in the production and the state-of-aggregation in the transformation as well as the target product to a large degree determine the strategy for upgrading. One avenue pursued for transforming glycerol is the gas-phase dehydration into acrolein, which has several potential applications. The molecule is already used as an intermediate today, based on propene oxidation traditionally over copper catalysts (supported or non-supported), and due to its two double bonds can react in a multitude of ways [3]. Possible products include allyl alcohol (1-propenol), pyridine, 3-picoline, acrylonitrile and acrylic acid.
The catalysts used for the dehydration reaction are mainly acidic and can be characterised according to the material used. In a previous paper by the authors [4] as well as a few available reviews [5,6,7] gives a good overview of the subject; the classes of catalysts used include: heteropoly acids and other supported inorganic acids [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26], zeolites [21, 27,28,29,30,31,32,33], mixed oxides [34,35,36,37,38,39,40,41], and sulphated zirconia [42, 43], all of which are solid acid materials. The acids usually contain or are promoted with P, W, S, Mo, and B, while ZrO2, Al2O3, SiO2, and TiO2 are supports of choice for heteropoly acid and mixed oxides. Glycerol conversion is normally high (80–100%) with yields maintained in the 60–80% range at high conversion rates [40]. The reaction is normally performed at moderate pressure and at medium to low temperature. Catalyst stability is an issue and carbon formation limits the time-on-stream.
There are several papers that investigate the deactivation of these kinds of catalysts under this reaction regime and carbon formation appears to be the general consensus. The carbon formation is suggested to depend on oligomer formation with hydroxyacetone as a precursor [31, 35]; indeed recycling of hydroxyacetone very much favours carbon formation [4]. Deleplanque et al. [35] imply that formation of olefins (yielding subsequent aromatics, oligomers and coke) is a possible parallel deactivation mechanism and in a separate paper Suprun et al. derived the deactivation to Brønstead acid sites [15]. The same researchers also found that the higher the temperature the lower the hydrogen-to-carbon quote was in the resulting deposits. Further, aside from the temperature, there is a marked lower carbon formation rate with increased water and oxygen content in the feed [26]. As a curiosity, it has also recently been reported that the formation of carbon on the catalyst increase the selectivity of WO3/TiO2 and heteropoly acid catalyst to acrolein [24, 25].
In previous research, the potential for the pore condensation of glycerol due to physical reasons was proposed as a means of increased deactivation rate of catalysts for glycerol dehydration [4]. This behaviour, higher carbon formation with smaller pores, has been reported in literature [33]. In this paper, this insight is used to thermally modify catalysts and thereby improving their lifetime and yield of acrolein. The morphological changes of the materials are reported as well as the initial activity and selectivity of the modified catalysts. Selected catalysts were also lifetime tested over many 100 s of hours. The target of the catalysts pre-treatment is to achieve a larger pore diameter to avoid pore condensation.
2 Experimental
This section describes the catalyst-testing procedure, along with the methods used for catalyst characterisation.
2.1 Catalyst Testing
The catalysts were investigated in two different test set-ups. The first one was used for the activity and selectivity measurements and the second one for the long-term deactivation experiments. In the activity and selectivity experimental setup, a carrier gas (hydrogen) was mixed with liquid 20% glycerol (99.5% Karlshamn Tefac AB) by weight in water before being vaporised in a pre-heater. The vaporised liquid/gas mixture was then passed through a stainless steel reactor with an inner diameter of 15 mm and an inlet temperature of 553 K, on to a condenser and gas separation step, and finally collected in a vessel. The stream of carrier gas (hydrogen) was regulated by a mass-flow controller to 1.67 cm3 s−1 and the stream of liquid to 0.005 g s−1 by a high-pressure liquid chromatography pump, resulting in a gas hourly space velocity of 500 h−1. The gas and liquid were mixed in a manifold before entering the pre-heater. The reactor and pre-heater were situated in two tube furnaces, controlled individually so that the inlet temperature of the catalyst bed was the preferred one. The catalyst bed temperature was monitored using two thermocouples, situated at the inlet and approximately 10 mm before the end of the catalyst bed. The reactor outlet pressure was set to 0.5 MPa absolute. The long-term testing rig was similar to the one described above and used the same conditions with respect to catalyst amount, flowrate and composition. The pressure drop over the reactor was monitored continuously. The reactor inner diameter was 17 mm for both systems.
A gas and a liquid sample were taken from time to time over the entire test period. The gas was analysed using a Varian CP-4900 two-channel micro gas chromatograph (equipped with one Mol Sieve 5A PLOT column and one PoraPlot column, with double TCD detectors). The liquid sample was analysed using a Varian CP-3800 gas chromatograph equipped with a 1177 split/splitless injector, a CP-wax 58/FFAP column, and a flame ionisation detector.
2.2 Material Preparation
The catalyst used was a commercially available catalyst that is 10% WO3 by weight supported on ZrO2 in the form of beads 1–2 mm in diameter (Daiichi KKK), pre-treated in different ways. The pretreatment performed was a heat treatment at different temperatures (500, 600, 700, 800, 850, 900 and 1000 °C) for 4 h.
2.3 Catalyst Characterisation
Several catalyst characterisation methods have been used to understand the changes in the catalyst morphology. These methods, presented here, are surface area measurement using nitrogen adsorption, mercury intrusion porosimetry, Raman spectroscopy as well as temperature programmed ammonia desorption. All catalysts were analysed for changes in their BET surface areas measured by the adsorption of nitrogen at liquid nitrogen temperature using a Micromeritics ASAP 2,400 instrument after degassing for 16 h at 473 K. Pore volume analysis was performed using the BJH method [44]. For calculating the adsorbed layer thickness (t value), The Halsey formula [45] was used. In the analysis, the desorption-isotherm was used.
Fourier transform (FT)-Raman spectra were recorded on a Bruker IFS 66 FT-IR spectrometer fitted with the Bruker FRA 106 FT-Raman add-on. A low-power, diode-pumped Nd:YAG laser, model DPY 301 (1064 nm) from Adlas, and a liquid-nitrogen-cooled, high-sensitivity model 403 germanium diode detector from Applied Detector Corp. were used. In the analysis, the laser power was 100 mW and the resolution was 4 cm−1. Every spectrum reported is an average of 800 scans. Mercury intrusion porosimetry (MIP) was performed using a Micromeretics AutoPore IV instrument at a maximum pressure of 414 MPa, using an assumed 130 degrees in contact or wetting angle, a surface tension of 485 mN m−1 and a density of 13 541.3 kg m−3 for the mercury.
The acidity of the catalyst was measured for the investigated catalysts using ammonia-TPD on a Micromeretics 2900. The catalyst sample was placed in a glass tube and degassed at 673 K for 1 h in helium atmosphere. Post degassing, the temperature was lowered to 323 K and the catalysts were saturated with ammonia using a gas mixture of 5% NH3 in He. Physisorbed ammonia was desorbed at 323 K by switching back to He before initiating the temperature ramp. The temperature ramp was set to 973 K with 10°/min ramp-rate.
3 Results
This section presents the results of the catalyst characterisation, along with the results of the catalyst testing. As a point of reference, to determine whether pore condensation are in place during the used test conditions, the Kelvin equation has been used (Eq. 1) in which R is the gas law constant, T the absolute temperature, P 0 the bulk saturated vapour pressure, P C the pressure at which pore condensation occurs, σ VL the gas–liquid surface tension derived from Gallant [46], θ the contact angle (12° used in the assessment) [47], n L and n G the bulk molar densities of the gas and liquid, respectively, and H the pore diameter. Using the equation with these assumptions for the testing conditions described above gives pore condensation in pores smaller than 45 Å (4.5 nm).
3.1 Characterisation
The aim of the thermal pre-treatment is to eliminate the pores below 45 Å, to avoid any pore condensation. To investigate the changes to the pore system, MIP was used. The results of these analyses may be viewed in Fig. 1. It is evident that during the first thermal treatments (400–800 °C) there is not much change to the catalyst. At 850 °C there is some change initiated, which is further continued with the treatment at 900 and 1000 °C. At 850 °C, the first peak in intrusion volume (small diameter pores) is moved upwards from the 100–120 Å range to 150–160 Å. Continued heat treatment at 900 °C shifts this peak to the 360–370 Å range and at 1000 °C to the 2000 Å range. For the 850 and 900 °C samples, there is also an increase in pore volume in the 500–700 Å range; for the 900 °C sample, there is also a significant shift upwards in pore size for this second peak compared to the fresh and low temperature treated samples. In the case of the 1000 °C, there are no other pores than the ones in the 2000 Å range.

The incremental pore volume of the heat-treated catalysts
The behaviour with a shift in the pore volume above 800 °C is confirmed by the BET analysis, Fig. 2. There is not much change in the first number of samples (400–700 °C), whilst there is a slight decrease in surface area (11%) at 800 °C. Thereafter the change in surface area is more pronounced, at 850 °C the decrease is almost 40%, at 900 °C it is 66% and at 1000 °C heat pre-treatment it is 93%.

The results from BET and TPR analysis
It is interesting to note that even though there is little change to the catalyst BET surface area for the first four catalyst samples, there are other changes. As evident from Fig. 2 the overall acidity increases with heat treatment up to the 600 °C sample, thereafter the specific acidity (mmol/g) as measured by ammonia adsorption is decreasing rather linearly with decreasing surface area.
In Fig. 3, the specific acid density (mmol/m2) and desorption maxima are reported for the catalysts investigated. It can be seen that initially, the specific acidity increases, but it then more or less levels out at about 2.05 × 10−6 mmol of NH3 per square meter of catalyst surface area. The final value for the 1000 °C sample is for obvious reasons less precise (lower values, higher uncertainty). At the same time, the temperature for maximum ammonia desorption is shifted downwards in temperature for the last three samples, above 800 °C. This implies that even though the specific acidity is constant or indeed slightly increasing for the samples above 800 °C, the acid strength of these sites decrease.

Specific acid density (mmol/m2) of the catalyst and desorption maxima for ammonia
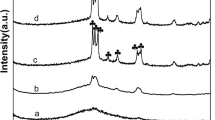
As may be viewed in Fig. 4, the Raman spectrum for the fresh catalyst and for the catalysts heat treated from 400 to 850 °C catalyst is consistent with that of monoclinic zirconia with strong intensity bands at 180, 188, 222, 305, 333, 345, 378, 475, 534, 569, 618, and 634 cm−1 [48]. The small shoulder at 850 cm−1 may be attributed to the W–O–W bond and the unresolved band at 900–990 cm−1 indicates W=O terminal bonds [8]. The presence of intensity bands at about 810 and 720 cm−1 are consistent with the formation of triclinic WO3 (tetragonal) for the catalyst samples heat-treated above 850 °C [49].

Raman-spectra of selected dehydration catalyst samples
3.2 Activity Measurements
The sintered catalysts were evaluated by activity tests in a micro-reactor, as a comparison the result from the fresh catalyst (not sintered), during a 20 h run, is given in Fig. 5.

Fresh non-sintered dehydration catalyst. Run at 553 K, 0.5 MPa and GHSV 500 h−1
Figures 6, 7 and 8 shows the results from the runs with the prepared catalysts sintered at 850, 900 and 1000 °C. The effect on the pore size distribution of the treatment can be seen in Fig. 1 above.

Catalyst sintered at 850 °C. Run at 553 K, 0.5 MPa and GHSV 500 h−1

Catalyst sintered at 900 °C. Run at 553 K, 0.5 MPa and GHSV 500 h−1

Catalyst sintered 1000 °C. Run at 553 K, 0.5 MPa and GHSV 500 h−1
To evaluate the results from the treated samples, some statistical evaluations have been done. Below in Fig. 9, the measured yields of acrolein as function of time on stream for the four samples, fresh and sinterd at 850, 900 and 1000 °C, are shown. The data are fitted with linear regression.

Linear regressions to the yield of acrolein as function of time
Straight line equation \({y}=k\, \times \,x+a\), where k is the slope and a is the intercept.
The linear fits to the yield of acrolein are shown in Fig. 9. The calculated regression coefficients R were tested for validity on the 95% level according to Eq. 2.
With the null hypothesis H0: No correlation exists, Table 1.
In Table 1, one can compare the test quantity t and the t-table value. In the case of the fresh sample (non-sintered), the test quantity t is smaller than the t-table value. This means that there is no significant correlation on the 95% level. In this case it indicates that the acrolein yield is a constant, it does not vary. The mean value is then 88.37 ± 0.41% for 95% confidence interval (CI).
In all other cases (the sintered samples) the test quantity is greater than the table value. This means that the null hypothesis “no correlation exists” can be discarded. Correlation exists in those cases, Table 2.
The decreases observed are probably not linear with time over a larger timespan. Catalysts usually have an initial deactivation phase and then the rate of deactivation levels off and the activity becomes constant. Therefore long-term tests were performed with selected catalysts. The catalyst sintered at 850 °C, were evaluated in a long-term activity test and compared to the fresh sample. In Fig. 10, the gas composition during 650 h is shown and it is obvious that the activity and selectivity are constant during the 650 h run.

Long term testing 20 wt% glycerol to acrolein catalyst heat-treated at 850 °C. Run at 553 K, 0.5 MPa and GHSV 500 h−1
In Fig. 11, the result from the sintered catalyst at 850 °C and for the un-sintered catalyst is shown. Not surprisingly, there is no correlation of yield to time in the statistic test of the regression coefficient. Meaning the measured values are constant over the timespan. The mean values are 75.0 ± 0.7% for the un-sintered catalyst and 87.6 ± 0.9% for the catalyst sintered at 850 °C. With those mean values and CI it is obvious that the values significantly differ.

Long term testing comparison of un-sintered catalyst and catalyst sintered at 850 °C. Run at 553 K, 0.5 MPa and GHSV 500 h−1
The limiting factor for the reaction is the coke formation in the reactor. This is manifested as an increased pressure drop over the reactor. In Fig. 12, the measured pressure drop (dP) over the reactor during the long-term test is shown.

Pressure drop as function of time for the heat-treated (850 °C) and fresh catalyst
As a comparison, the recorded pressure drop from the long-term test with the fresh (unsintred) catalyst (from Fig. 12) is shown in the same figure. It takes longer time for the sintered catalyst to reach the same pressure drop as the unsintered catalyst. This means that the time on stream before regeneration has been prolonged for the treated catalyst. This is probably due to the higher selectivity to acrolein obtained by the sintering. If the selectivity towards acrolein is improved, the formation of by-products is decreased. This leads to a longer availability on stream.
4 Discussion
The goal of this work has been to remove pores with diameter <45 Å in an attempt to improve the performance in the dehydration reaction of glycerol into acrolein. It has been shown in this work, that it is possible to change the pore structure of the WO3/ZrO2 catalyst by thermal sintering in a controlled way (Fig. 1), without changing the morphology of the catalyst in a radical way. Up to a temperature of 850 °C, the WO3 phase remains in the monoclinic phase, but at 900 °C it changes into the triclinic phase (Fig. 4). The specific amount of acidity (mmol/g), measured by NH3 adsorption, decreases when the BET-surface of the catalyst decreases (Fig. 4). However, per m2 of the catalyst, the acidity of the thermally treated catalyst increases compared with the fresh one. Above 850 °C, the acidic strength, measured as peak desorption temperature of ammonia, starts to decrease (Fig. 3).
After the thermal treatment, the initial yields (up to 20 h) differs between the samples 900 °C > 850 °C > Fresh > 1000 °C. The reason for this is an improved selectivity for acrolein and deceased selectivity for mainly hydroxyacetone for the samples treated at 850 and 900 °C. However, when the catalyst is sintered at too high temperature, at 1000 °C, this effect is lost. While the fresh catalyst seems to have an initial stable and constant conversion, or at least the rate-of-deactivation is below the standard deviation in the measurements. All of the thermally treated samples have a significantly decreasing yield in the short time tests (0–20 h). It is not uncommon that fresh catalyst has an initial deactivation period and then the activity level out and become stable.
So the initial deactivation is introduced by the thermal treatment. If water desorption from the catalyst surface is a rate determine step in the reaction, a dehydrated surface will show a higher reaction rate initially than a fully hydrated one. When the water coverage increases, the rate will drop until it reaches steady state and explain the initial behaviour of the catalysts. In the long-term tests, up to 600 h, run in the fully automated long-term test rig, the starting yield was somewhat lower than in the short term run, 75.0 ± 0.7% for the fresh and 87.6 ± 0.9% for the sample sintered at 850 °C (Fig. 11). This is most likely due to the small differences in the set-ups, possible variations in temperature profiles etc. However, no significant drop in the yield could be detected for either of the samples during the run for up to 600 h. The performance of the thermally treated samples, below 1000 °C, is significantly improved with respect to the acrolein yield. The increase in acrolein yield with increasing coking, as previously reported [24, 25], cannot be confirmed in this study, at least not on this timescale; perhaps this phenomenon is more of an initial change.
From the characterization of the samples, we know that the BET surface, the specific acidic amount and the acidic strength decreases due to the thermal treatment. However, the number of acidic sites per m2 of the catalyst increase. If more than one site is required for the dehydration reaction of glycerol, a higher density of acidic sites on the surface could be favourable, or it may be the decrease in acidic-strength that is positive for the glycerol to acrolein reaction.
Decreasing the hydroxyacetone selectivity should give less coke formation in the catalyst, validated in earlier studies by co-feeding hydroxyacetone with the glycerol [4]. To investigate the carbon formation, the pressure drops over the catalytic beds was measured during the long-term test run. It was difficult to obtain stable readings, and other factors than coking could affect the result for instance packing of bed during the run or fragmenting of the catalyst, but the results in Fig. 12 indicates an improved time-on-stream with the catalyst heat-treated at 850 °C compared to the unsintered catalyst.
5 Conclusions
In conclusion, the pre-treatment of tungsta/zirconia catalysts by heat treatment changes the performance of the catalysts. The heat treatment changes the initial deactivation behaviour and the acrolein selectivity is increased initially, most likely due to a difference in surface-water coverage. The heat treatment also influences the catalyst morphology such that heat treatment (at temperatures above 800 °C) result in a catalyst with lower specific surface area, more open pore structure (larger pores), lower absolute acidity and acid strength, but higher specific acid density (mmol/m2). There is also a recrystallization of the tungsta-phase from monoclinic to triclinic between 850 and 900 °C. The catalyst with larger pore system, with no pores at 45 Å or below, also performs better in long-term testing compared to the non-heat treated sample. This indicate that pore condensation may be a contributing factor in catalyst deactivation, but other factors such as acid strength and specific acidity may also influence the behaviour.
References
Werpy TA, Holladay JE, White JF (2004) Top value added chemicals from biomass: I. Results of screening for potential candidates from sugar and synthesis gas. Department of Energy, Richland
Pagliaro M, Rossi M (2010) The future of glycerol. Royal Society of Chemistry, London
Arpe HJ, Hawkins S (2010) Industrial organic chemistry. Wiley, Hoboken
Hulteberg C, Leveau A, Brandin J (2013) Pore condensation in glycerol dehydration. Top Catal 56(9–10):813–821
Katryniok B, Paul S, Belliere-Baca V, Rey P, Dumeignil F (2010) Glycerol dehydration to acrolein in the context of new uses of glycerol. Green Chem 12(12):2079–2098
Katryniok B, Paul S, Capron M, Dumeignil F (2009) Towards the sustainable production of acrolein by glycerol dehydration. ChemSusChem 2(8):719–730
Dam JT, Hanefeld U (2011) Renewable chemicals: dehydroxylation of glycerol and polyols. ChemSusChem 4(8):1017–1034
Chai SH, Wang HP, Liang Y, Xu BQ (2009) Sustainable production of acrolein: preparation and characterization of zirconia-supported 12-tungstophosphoric acid catalyst for gas-phase dehydration of glycerol. Appl Catal A 353(2):213–222
Tsukuda E, Sato S, Takahashi R, Sodesawa T (2007) Production of acrolein from glycerol over silica-supported heteropoly acids. Catal Commun 8 (9):1349–1353
Shen L, Feng Y, Yin H, Wang A, Yu L, Jiang T, Shen Y, Wu Z (2011) Gas phase dehydration of glycerol catalyzed by rutile TiO2-supported heteropolyacids. J Ind Eng Chem 17(3):484–492
Atia H, Armbruster U, Martin A (2008) Dehydration of glycerol in gas phase using heteropolyacid catalysts as active compounds. J Catal 258(1):71–82
Kim YT, Jung K-D, Park ED (2011) Gas-phase dehydration of glycerol over silica–alumina catalysts. Appl Catal B 107(1–2):177–187
Atia H, Armbruster U, Martin A (2011) Influence of alkaline metal on performance of supported silicotungstic acid catalysts in glycerol dehydration towards acrolein. Appl Catal A 393(1–2):331–339
Suprun W, Lutecki M, Papp H (2011) TPD-TG-MS investigations of the catalytic conversion of glycerol over MOx-Al2O3-PO4 catalysts. Chem Eng Technol 34 (1):134–139
Suprun W, Lutecki M, Haber T, Papp H (2009) Acidic catalysts for the dehydration of glycerol: activity and deactivation. J Mol Catal A 309(1–2):71–78
Suprun W, Lutecki M, Gläser R, Papp H (2011) Catalytic activity of bifunctional transition metal oxide containing phosphated alumina catalysts in the dehydration of glycerol. J Mol Catal A 342–343(0):91–100
Ágnes Z, Péter B, Mónika F, Ferenc N (2010) Catalytic dehydration of glycerol under mild condition: an environmentally benign acrolein production. J Environ Prot 1(02):201–205
Alhanash A, Kozhevnikova EF, Kozhevnikov IV (2010) Gas-phase dehydration of glycerol to acrolein catalysed by caesium heteropoly salt. Appl Catal A 378(1):11–18
Munshi MK, Lomate ST, Deshpande RM, Rane VH, Kelkar AA (2010) Synthesis of acrolein by gas-phase dehydration of glycerol over silica supported Bronsted acidic ionic liquid catalysts. J Chem Tech Biotechnol 85(10):1319–1324
Katryniok B, Paul S, Capron M, Lancelot C, Belliere-Baca V, Rey P, Dumeignil F (2010) A long-life catalyst for glycerol dehydration to acrolein. Green Chem 12(11):1922–1925
Chai S-H, Wang H-P, Liang Y, Xu B-Q (2007) Sustainable production of acrolein: investigation of solid acid-base catalysts for gas-phase dehydration of glycerol. Green Chem 9(10):1130–1136
Chai S-H, Wang H-P, Liang Y, Xu B-Q (2008) Sustainable production of acrolein: gas-phase dehydration of glycerol over 12-tungstophosphoric acid supported on ZrO2 and SiO2. Green Chem 10(10):1087–1093
Shiju NR, Brown DR, Wilson K, Rothenberg G (2010) Glycerol valorization: dehydration to acrolein over silica-supported niobia catalysts. Topics Catal 53(15):1217–1223
Dalil M, Carnevali D, Edake M, Auroux A, Dubois J-L, Patience GS (2016) Gas phase dehydration of glycerol to acrolein: coke on WO3/TiO2 reduces by-products. J Mol Catal A 421:146–155
Dalil M, Edake M, Sudeau C, Dubois J-L, Patience GS (2016) Coke promoters improve acrolein selectivity in the gas-phase dehydration of glycerol to acrolein. Appl Catal A 522:80–89
Martinuzzi I, Azizi Y, Zahraa O, Leclerc J-P (2015) Deactivation study of a heteropolyacid catalyst for glycerol dehydration to form acrolein. Chem Eng Sci 134:663–670
Kim YT, Jung K-D, Park ED (2011) A comparative study for gas-phase dehydration of glycerol over H-zeolites. Appl Catal A 393(1–2):275–287
Yoda E, Ootawa A (2009) Dehydration of glycerol on H-MFI zeolite investigated by FT-IR. Appl Catal A 360(1):66–70
Kim YT, Jung KD, Park ED (2010) Gas-phase dehydration of glycerol over ZSM-5 catalysts. Microporous Mesoporous Mater 131(1–3):28–36
de Oliveira AS, Vasconcelos SJS, de Sousa JR, de Sousa FF, Filho JM, Oliveira AC (2011) Catalytic conversion of glycerol to acrolein over modified molecular sieves: activity and deactivation studies. Chem Eng J 168(2):765–774
Corma A, Huber GW, Sauvanaud L, Connor P (2008) Biomass to chemicals: catalytic conversion of glycerol/water mixtures into acrolein, reaction network. J Catal 257(1):163–171
Vieira LH, Carvalho KTG, Urquieta-González EA, Pulcinelli SH, Santilli CV, Martins L (2016) Effects of crystal size, acidity, and synthesis procedure on the catalytic performance of gallium and aluminum MFI zeolites in glycerol dehydration. J Mol Catal A 422:148–157
Rodrigues MV, Vignatti C, Garetto T, Pulcinelli SH, Santilli CV, Martins L (2015) Glycerol dehydration catalyzed by MWW zeolites and the changes in the catalyst deactivation caused by porosity modification. Appl Catal A 495:84–91
Lauriol-Garbey P, Postole G, Loridant S, Auroux A, Belliere-Baca V, Rey P, Millet JMM (2011) Acid-base properties of niobium-zirconium mixed oxide catalysts for glycerol dehydration by calorimetric and catalytic investigation. Appl Catal B 106(1–2):94–102
Deleplanque J, Dubois JL, Devaux JF, Ueda W (2010) Production of acrolein and acrylic acid through dehydration and oxydehydration of glycerol with mixed oxide catalysts. Catal Today 157(1–4):351–358
Chai S-H, Wang H-P, Liang Y, Xu B-Q (2007) Sustainable production of acrolein: gas-phase dehydration of glycerol over Nb2O5 catalyst. J Catal 250(2):342–349
Lauriol-Garbey P, Millet JMM, Loridant S, Belliere-Baca V, Rey P (2011) New efficient and long-life catalyst for gas-phase glycerol dehydration to acrolein. J Catal 281(2):362–370
Tao L-Z, Chai S-H, Zuo Y, Zheng W-T, Liang Y, Xu B-Q (2010) Sustainable production of acrolein: acidic binary metal oxide catalysts for gas-phase dehydration of glycerol. Catal Today 158(3–4):310–316
Ulgen A, Hoelderich W (2009) Conversion of glycerol to acrolein in the presence of WO3/ZrO2 catalysts. Catal Lett 131(1):122–128
Ulgen A, Hoelderich WF (2011) Conversion of glycerol to acrolein in the presence of WO3/TiO2 catalysts. Appl Catal A 400(1–2):34–38
Lauriol-Garbay P, Millet JMM, Loridant S, Bellière-Baca V, Rey P (2011) New efficient and long-life catalyst for gas-phase glycerol dehydration to acrolein. J Catal 280(1):68–76
Cavani F, Guidetti S, Marinelli L, Piccinini M, Ghedini E, Signoretto M (2010) The control of selectivity in gas-phase glycerol dehydration to acrolein catalysed by sulfated zirconia. Appl Catal B 100(1–2):197–204
Cavani F, Guidetti S, Trevisanut C, Ghedini E, Signoretto M (2011) Unexpected events in sulfated zirconia catalyst during glycerol-to-acrolein conversion. Appl Catal A 409–410:267–278
Barrett EP, Joyner LG, Halenda PP (1951) The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J Am Chem Soc 73(1):373–380
Halsey G (1948) Physical adsorption on non-uniform surfaces. J Chem Phys 16(10):931–937
Gallant RW (1967). Hydrocarb Process 46(5):201–215
Ma J, Jing G, Chen S, Yu D (2009) Contact angle of glycerol nanodroplets under van der Waals force. J Phys Chem C 113(36):16169–16173
Zyuzin DA, Cherepanova SV, Moroz EM, Burgina EB, Sadykov VA, Kostrovskii VG, Matyshak VA (2006) X-ray, Raman and FTIRS studies of the microstructural evolution of zirconia particles caused by the thermal treatment. J Solid State Chem 179(10):2965–2971
Salje E (1975) Lattice dynamics of WO3. Acta Crystallogr Sect A 31(3):360–363
Acknowledgements
Mrs. Birgitta Lindén for performing BET and MIP analysis. The financial support from Energiforsk and the Swedish knowledge centre for renewable transportation fuels are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hulteberg, C., Leveau, A. & Brandin, J.G.M. Pore Condensation in Glycerol Dehydration: Modification of a Mixed Oxide Catalyst. Top Catal 60, 1462–1472 (2017). https://doi.org/10.1007/s11244-017-0827-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-017-0827-8