
Abstract
The catalytic dehydration of glycerol to acrolein is investigated over silica-supported niobia catalysts in a continuous fixed-bed gas-phase reactor. Various supported niobia catalysts are prepared and characterized using surface analysis and spectroscopic methods (XRD, UV–Vis, XPS, N2 adsorption), as well as with ammonia adsorption microcalorimetry. Good results are obtained with initial glycerol conversions of over 70% and with 50–70% selectivity to acrolein. We investigate the influence of changing the catalyst acid strength by varying the niobia content and catalyst calcination temperature. Glycerol conversion and acrolein selectivity depend on the surface acid strength. Catalyst deactivation by coking is also observed, but simple oxidative treatment in air restores the activity of the catalysts completely.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The search for catalytic processes targeting renewable raw materials for producing bulk chemicals is a ‘hot topic’. Aiming at sustainable and ‘green’ processes, chemists must keep in mind the big picture, i.e. an overview of not only which raw materials are used, but where they’re coming from and how do they relate to adjacent processes. Glycerol is a good example. It is a by-product of biodiesel synthesis from vegetable oil and animal fat (triglycerides), and though the amount of glycerol per tonne of biodiesel is small (ca. 1:10 ratio), the large tonnage of biodiesel for fuels means sufficient supply of glycerol for chemicals. In 2008, the total EU biodiesel production was 7.76 MMT [1]. This must increase considerably to meet the 4% target set by the EC [2]. Thus, the supply of glycerol in the coming decades is ensured, and with new heterogeneous catalytic processes for esterification and transesterification, the quality of this glycerol will only improve.
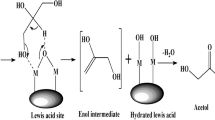
One important option for using this glycerol is converting it catalytically to acrolein via double-dehydration (Scheme 1). Such a process would offer a sustainable alternative to the present acrolein manufacturing technology that starts from petroleum-derived propylene. Acrolein is a key bulk chemical, produced on million-ton scale worldwide. It is used in making various products, including acrylic acid, acrylic esters, absorbent polymers and detergents [3].

Dehydration of glycerol to acrolein
To date, works involve dehydration of glycerol in either liquid or gas-phase using zeolites, oxides, sulfates, phosphates, heteropoly acids, and supported phosphoric acid [4–16]. Acrolein has also been prepared from glycerol using subcritical and supercritical water [17–19]. None of these processes are applied on a commercial scale.
Since glycerol is usually produced as a mixture with water, we wanted to use the glycerol–water mixture directly instead of pure glycerol for producing acrolein, requiring a catalyst with water-tolerant properties in addition to favourable acidic properties [20, 21]. Niobia was previously used as a solid acid catalyst in reactions that require water-tolerance [22, 23], so we selected supported niobia as the catalyst for the dehydration of glycerol to acrolein. As described herein, we have prepared various silica-supported niobia catalysts, characterised them and tested them in gas-phase glycerol dehydration.
2 Experimental Section
2.1 Materials and Instrumentation
Silica gel (Merck) was used as support (particle size = 0.063–0.200 mm). Before impregnation, silica gel was calcined at 400 °C. After calcination, the surface area was 480 m2 g−1 and pore volume was 0.79 cm3 g−1. Other chemicals were purchased from Aldrich and used as received. The niobia precursor used was ammonium niobate (V) oxalate hydrate, C4H4NNbO9·xH2O (99.99%, Aldrich). The gases were purchased from BOC. Powder X-ray diffraction patterns were collected on a Bruker diffractometer using Cu Kα radiation. The N2 adsorption–desorption isotherms were measured at 77 K on a Micromeritics ASAP-2000 after evacuation at 473 K for 5 h. The surface areas and the average pore sizes were calculated by the BET and BJH methods, respectively. XPS measurements were performed using a Kratos AXIS HSi instrument equipped with a charge neutraliser and Mg Kα X-ray source. Spectra were recorded at normal emission using an analyser pass energy of 40 eV, X-ray power of 144 W and were energy referenced to the valence band and adventitious carbon. Survey scans were recorded at 160 eV pass energy. Spectra were Shirley background-subtracted across the energy region and deconvoluted using Casa XPS Version 2.3.15. The TG analysis was carried out using a thermal-analyzer (Mettler-Toledo) at a heating rate of 10 °C min−1. UV–Vis spectra were collected on Perkin-Elmer λ 35 spectrometer, using a labsphere reflectance spectroscopy accessory. The system used for ammonia adsorption flow calorimetry has been described previously [24, 25]. Briefly, it is based on a Setaram 111 DSC with an automated gas flow and switching system, with a mass spectrometer (Hiden HPR20). After activation of the sample (20–30 mg) at 150 °C under a dried helium flow, 1 mL pulses of the probe gas (1% ammonia in helium) at atmospheric pressure were injected at regular intervals into the carrier gas stream from a gas sampling valve. The net amount of ammonia irreversibly adsorbed from each pulse was determined by comparing the MS signal with that recorded through a control experiment with a blank sample tube. The net heat released by each pulse was calculated from the thermal DSC curve.
2.2 Procedure for Catalyst Preparation
All catalysts were prepared by impregnating the silica support with calculated amounts of an aqueous solution of ammonium niobium oxalate [C4H4NNbO9·xH2O] to achieve the required loadings. The samples were then dried at 80 °C for 12 h and calcined at various temperatures in static air for 4 h. The details of catalysts are provided in Table 1.
2.3 Procedure for Catalyst Testing
The gas-phase dehydration reaction of glycerol to acrolein was carried out under atmospheric pressure in a vertical fixed-bed quartz reactor. A constant volume of catalyst was charged in the middle section of the reactor, with quartz wool packed in both ends. The reactor was also packed with ceramic beads above the catalyst bed in order to preheat and to vaporise the feed. The catalyst was then pretreated at 400 °C for 1 h in a flow of nitrogen (60 mL min−1). The temperature was then lowered to the required reaction temperature and the reaction feed, an aqueous solution containing 30 wt% glycerol, was fed into the reactor by a syringe pump. The reaction products were condensed in an ice-water trap and collected hourly for analysis on a Perkin Elmer Clarus 500 GC using a 50 m BP5 capillary column and an FID. The products collected during the first hour of the reaction were abandoned due to poor material balance.
3 Results and Discussion
3.1 Catalyst Preparation and Characterization
The catalysts were prepared by impregnating an aqueous solution of ammonium niobium oxalate onto silica. After the impregnation, the catalysts were dried and calcined in air. Table 1 shows the catalyst details and characterization data. Figure 1 shows the X-ray diffraction patterns of silica-supported niobia catalysts calcined at various temperatures. For samples with niobia loading lower than 40 wt%, only a broad peak was observed. This was the case also for the sample containing 40% niobia, which was calcined at lower temperatures. This peak, centered at 22.6°, represents the orthorhombic (T) phase of niobium pentoxide [26]. For catalyst 8 (see Table 1 for details), more peaks were observed which can be assigned to characteristic reflections of T-Nb2O5. Pure Nb2O5 undergoes crystallisation at lower temperatures, hence the XRD of the present samples show that higher temperatures are required for the crystallisation of niobium pentoxide when supported on silica. This indicates a high dispersion of niobia and stronger interaction between the supported niobia species and silica. Such interactions are frequently observed for supported metal oxides [27].

X-ray diffraction patterns of some of the supported niobia catalysts. See Table 1 for the details of catalysts
Table 1 gives the textural properties derived from nitrogen physisorption isotherms. Surface areas and pore volumes decreased with increasing the calcination temperature. The average pore diameter was 4.3–5.0 nm, decreasing for higher niobia loadings. The thermogravimetric profiles of 20 and 40% Nb2O5 samples showed that the decomposition of ammonium niobium oxalate occurred between 200 and 300 °C, with increased mass losses at higher Nb content (Fig. 2).

TG analysis of silica-supported niobia catalysts with 20 and 40% Nb2O5 loading
The acidities were evaluated by ammonia adsorption microcalorimetry. In this method, the heat of NH3 adsorption is a direct measure of the total acid strength, and the surface coverage of NH3 gives an idea of the number of acid sites. The method, however, cannot distinguish Brønsted/Lewis sites. The initial heat of adsorption on silica support was ~37 kJ mol−1 while the niobia containing samples showed a three-fold increase. This indicates the formation of new surface sites with higher acid strength. Since the Nb–O bond has a higher ionic character, Lewis acid sites could be generated when Nb2O5 is incorporated into the silica matrix. The higher electronegativity of Nb (V) compared to Si (IV) could also cause the formation of Brønsted acid sites by weakening of the O–H bond. The strength of acid sites increased with increasing niobia loading up to 20 wt%, and decreased thereafter (Fig. 3). The acid strength decreased at higher calcination temperatures, as expected. Since higher temperatures lead to the formation of niobia particles, as indicated by XRD, we assume that the acid strength is higher when the niobia phase is well dispersed. This agrees with the decreased acid strength of catalyst 5 for which the niobia peaks are better defined, compared to catalyst 4 (Fig. 4).

NH3 adsorption microcalorimetry data as a function of niobia loading. See Table 1 for catalyst details

NH3 adsorption microcalorimetry data of sample containing 20% niobia as a function of calcination temperature. See Table 1 for catalyst details
Samples were analysed by XPS, probing surface composition and Nb oxidation state. Representative Nb 3d XP spectra are shown in Fig. 5 for the sample containing 20% Nb2O5 calcined at 400 °C (catalyst 4). We see a broad set of spin/orbit split doublets for the 3d5/2 and 3d3/2 states at 207.9 and 210.5 eV. The 3d5/2 state can be deconvoluted into two components, with binding energies of 207.7 and 208.7 eV, respectively, in good agreement with the presence of Nb5+, with the 3d5/2 for Nb2O5 expected at 207.6 eV [25]. The high binding energy component at 208.7 eV may reflect perturbation of the Nb2O5 which is in direct contact with the support, i.e. at the interface. Such broadening effects were observed previously for Mo–Nb2O5/SiO2 catalysts [28]. Following calcination at 800 °C (catalyst 7) there is a reduction in the Nb 3d intensity of the 207.6 eV component and further broadening of the doublet with a new lower binding energy state at 207.4 eV evolving. Nb, NbO and NbO2 are expected at 202.2, 203.7 and 205.7 eV respectively, so it is unlikely that this new low binding energy state pertains to reduction of Nb2O5. We think that high-temperature calcination causes sintering of the Nb2O5 crystallites, thus shifting the binding energy. The contribution from the interfacial Nb2O5 remains unchanged, suggesting that it is stable. The surface compositions of the materials determined from XPS show a slight decrease in the Nb/Si atomic ratio from 0.04 to 0.03. This could be due to Nb loss or Nb dissolution into the silica matrix during calcination.

Nb 3d X-ray photoelectron spectra of the sample containing 20% Nb2O5
The oxidation state of Nb was further confirmed by UV–Vis DRS spectroscopy (Fig. 6). The spectra are dominated by intense bands corresponding to ligand-to-metal charge-transfer transitions (LMCT from O2− to Nb5+). However, the type of coordination, that is, tetrahedral (230 nm) or octahedral Nb species (≈250 nm), could not be distinguished from these broad bands.

UV–Vis DRS spectra of bulk niobia and catalyst 5
3.2 Catalyst Testing
We tested the catalysts for the dehydration of glycerol in a gas-phase fixed bed down-flow reactor. The results show that silica-supported niobia catalysts are active for the catalytic dehydration of glycerol. Catalysts exhibited very high initial glycerol conversions, and almost 100% conversion was obtained with catalyst 4 after 2 h. The major product was acrolein. With silica support alone, very low conversion of glycerol and poor selectivity to acrolein was observed. This indicates that the acidic sites generated by the supported niobia are the actual active species. The byproducts produced during the reaction were 1-hydroxyacetone, acetone, acetaldehyde, allyl alcohol, propionaldehyde and acetic acid, all of which were formed with low selectivities. A number of other byproducts which remain unidentified also were produced in very minor amounts (<2%).
The glycerol conversion increases with niobia loading, reaching a maximum at 20% (Fig. 7a). The selectivity to acrolein also follows the same trend (Fig. 7b). Note that calcination at higher temperatures such as 800 °C decreased both glycerol conversion and selectivity to acrolein (Fig. 8).

Glycerol conversion (a) and acrolein selectivity (b) as a function of time on stream for catalysts with different niobia loading. Reaction Temperature: 320 °C, WHSV = 80 h−1 and feed = 30% solution of glycerol in water. See Table 1 for catalyst details

Glycerol conversion and acrolein selectivity as a function of time on stream for 20Nb2O5–SiO2 calcined at 400 °C (filled square, open square), 600 °C (filled triangle, open triangle) and 800 °C (filled circle, open circle). Solid symbols indicate glycerol conversion and open symbols indicate acrolein selectivity. Reaction Temperature: 320 °C, WHSV = 80 h−1 and feed = 30% solution of glycerol in water
Comparing these activity results with the acid properties of catalysts evaluated by ammonia adsorption microcalorimetry (Figs. 3, 4) brings out the relation between the acid strength and the acrolein yield. Higher acidic strength is favourable for a higher acrolein yield for this set of catalysts. This can be better understood by Fig. 9, in which heat of adsorption and acrolein formation rate are plotted together. The mass-specific catalytic rates were obtained by normalizing the glycerol consumption and acrolein formation data according to the weight of each catalyst. As can be seen from the figure, these rates were linearly related with the heat of adsorption of ammonia which means that higher acidic strength favours higher yield of acrolein for this set of catalysts.

Relationship between acrolein formation rate (obtained at time on stream = 9–10 h) and heat of adsorption for silica-supported niobia catalysts
Though the initial conversions were very high, for all catalysts, conversion of glycerol decreased rapidly with time-on-stream (TOS) as can be observed from Fig. 7, indicating a fast deactivation. However, there was no significant change in the selectivity pattern with TOS and acrolein was the major product. Since the catalysts are expected to possess water tolerant properties, the most probable reason for the deactivation is coking. To confirm this, the catalysts were removed after reaction and were analysed by TG in N2 flow. There was a weight loss on the TG curve between 350 and 650 °C due to burn off of the carbon deposits (Fig. 10). This indicates that the deactivation is indeed mainly due to the coking. The fact that catalysts deactivate in spite of the presence of a large amount of steam during the reaction emphasizes high probability of the side reactions leading to the formation of carbonaceous products. Water tolerance is not sufficient to avoid the deactivation and acid properties need to be fine tuned for this purpose. Nonetheless, the catalysts could be fully regenerated by recalcination of the used catalyst at around 500 °C for 5 h in flowing air (30 mL min−1).

TG analysis in N2 flow of the catalyst 4 removed after reaction
4 Conclusions
Silica-supported niobia catalysts showed significant activity in the catalytic dehydration of glycerol to acrolein in the presence of water. The conversion of glycerol and selectivity to acrolein depends on the niobia loading and calcination temperature. The total acidity was changed depending on the niobia loading and calcination temperature of the catalysts. Catalyst performance was strongly influenced by the total acidity. Deactivation due to carbon deposition was observed for all the catalysts, but an oxidative treatment in air at temperatures around 500 °C sufficed to recover both acidity and activity.
References
Statistics of European Biodiesel Board. http://www.ebb-eu.org/stats
Official Journal of the European Union, L140/6, 05-06-2009
Corma A, Huber GW, Sauvanaud L, O’Connor P (2008) J Catal 257:163
Haas T, Arntz D, Klenk H, Girke W (1994) Pat. EP. 19,930,116,792
Dubois J-L, Duquenne C, Hölderich W, Kervenal J (2006) Pat. WO 2006/087084 A2
Tsukuda E, Sato S, Takahashi R, Sodesawa T (2007) Catal Commun 8:1349
Chai S, Wang H, Liang Y, Xu B (2007) Green Chem 9:1130
Atia H, Ambruster U, Martin A (2008) J Catal 258:71
Suprun W, Lutecki M, Haber T, Papp H (2009) J Mol Catal A 309:71
Zhou C, Beltramini JN, Fana Y, Lu GQ (2008) Chem Soc Rev 37:527
Jerome F, Pouilloux Y, Barrault J (2008) ChemSusChem 1:586
Chai S, Wang H, Liang Y, Xu B (2007) J Catal 250:342
Wang F, Dubois J, Ueda W (2009) J Catal 268:260
Chai S, Wang H, Liang Y, Xu B (2009) Appl Catal A 353:213
Peterson TH, Holladay JE, Zacher AH, Gray MJ, Werpy TA, White JF (2009) US20090054694
Peterson TH, Zacher AH, Gray MJ, White JF, Holladay JE, Werpy TA (2009) US20090054538
Ott L, Bicker M, Vogel H (2006) Green Chem 8:214
Ramayya S, Brittain A, DeAlmeida C, Mok W, Antal MJ (1987) Fuel 66:1364
Watanabe M, Iida T, Aizawa Y, Aida TM, Inomata H (2007) Bioresour Technol 98:1285
Gordillo A, Durán Pachón L, de Jesus E, Rothenberg G (2009) Adv Synth Catal 351:325–330
Kiss AA, Dimian AC, Rothenberg G (2006) Adv Synth Catal 348:75
Ziolek M (2003) Catal Today 78:47
Tanabe K, Okazaki S (1995) Appl Catal A 133:191
Shiju NR, AnilKumar M, Hoelderich WF, Brown DR (2009) J Phys Chem C 113:7735
Shiju NR, Williams HM, Brown DR (2009) Appl Catal B 90:451
Nowak I, Ziolek M (1999) Chem Rev 99:3603
Shiju NR, Anilkumar M, Mirajkar SP, Gopinath CS, Rao BS, Satyanarayana CV (2005) J Catal 230:484
Damyanova S, Dimitrov L, Petrov L, Grange P (2003) Appl Surf Sci 214:68
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shiju, N.R., Brown, D.R., Wilson, K. et al. Glycerol Valorization: Dehydration to Acrolein Over Silica-Supported Niobia Catalysts. Top Catal 53, 1217–1223 (2010). https://doi.org/10.1007/s11244-010-9566-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-010-9566-9