
Abstract
New methodologies that enable palladium catalyzed cross-coupling reactions to be performed under environmentally benign conditions (in water and/or at room temperature) have been developed. Described approaches involve in situ activation of carbon–halogen or carbon–hydrogen bonds using zinc metal, or cationic palladium, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
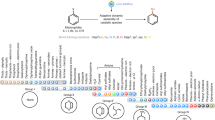
Cross-couplings catalyzed by transition metals historically rely on a reactive electrophile into which a transition metal inserts, and a reactive nucleophile that transfers an organic fragment to the transition metal center. Reductive elimination finally delivers the product. Over time, milder and more selective conditions for cross-coupling reactions have been developed [1–3]. More active palladium catalysts have emerged by virtue of new ligands of broad structural variation, such as phosphines, phosphoramidites, NHCs (N-heterocyclic carbenes), diamines, P,N–, N,O–, etc. [4, 5]. Different nucleophiles (e.g., organoboron, organotin, organozinc, and organomagnesium reagents, amines, alkenes, alkynes, etc.) define a variety of “name” reactions routinely used in organic synthesis today. Insofar as green chemistry is concerned, however, many of these reactions leave considerable room for improvement. Notwithstanding the fact that each involves “catalysis” by transition metals, which is an inherently green characteristic [6], high temperatures, potentially toxic organic solvents, highly reactive organometallic reagents, and use of super-stoichiometric levels of additives clearly present challenges to designing alternative processes which are far more environmentally benign. Prevention of waste, atom economy, safer solvents, energy efficiency, limited workup and derivatization, and minimal by-product formation: all are worthy properties that are associated with the methodologies that follow herein, in line with the principles of green chemistry (Fig. 1) [7].

Organometallic cross-couplings of the future: fewer steps, greener conditions
2 Cross-Couplings in Water at Room Temperature: No Preformed Organometallic
From a practical standpoint, the more reactive the nucleophile in cross-coupling reactions the greater the level of care with which it has to be prepared to avoid moisture, and handled in an inert atmosphere to minimize oxidation. Protioquenching and/or reagent/ligand oxidation can be major impediments to the use of such reagents. Moreover, low functional group tolerance associated, in particular, with Grignard reagents as in Kumada couplings [8, 9], may necessitate protecting group chemistry, adding steps (protection, deprotection) to the sequence and usually reducing overall yields. Carbon–carbon bond formation based on cross-couplings that entirely avert prior formation of a nucleophile would be advantageous. The challenge is that two (or more) electrophilic species must co-exist in one pot, where one ultimately serves as the in situ-derived nucleophile. Can conditions be found such that competition between two electron-rich metals for different electrophiles is averted, achieving a net umpolung/coupling? [10]. If prior formation of a stoichiometric organometallic species is unnecessary, can the reaction medium be environmentally friendly, perhaps even water? [11].
We recently described selective zinc metal insertion into sp3 carbon–halogen bonds that takes place in the presence of palladium(0), the latter preferentially inserting into sp2 aryl halides also present in the pot (Scheme 1) [12]. Alkyl-substituted arenes are formed, while homocoupling commonly seen with zinc-based and other organometallic reagents is suppressed by in situ generation of RZnX followed by transmetalation to palladium. Nanometer-size micelle-forming surfactants, in water as the only medium, presumably further slow down competitive protioquenching of newly forming organozinc species prior to transmetalation. Reactions “on water” [13] (in the absence of nanoparticles) do not give satisfactory yields of desired alkylated arenes. A crucial component in this process is diamine TMEDA (N,N′-tetramethylethylenediamine), its role seemingly multifaceted in that it (a) may activate the zinc surface prior to oxidative addition, akin to the role played by NH4Cl [14, 15]; (b) stabilizes the catalytic amounts of newly formed primary organozinc species by ligation, and (c) adds lipophilicity to the organometallic further enhancing its migration into the hydrophobic micellar interior. In the case of secondary alkyl iodides that are far more prone to Zn insertion, a change in diamine (to N,N′-tetraethylethylenediamine) was required to achieve good yields [13].

Zn-mediated, Pd-catalyzed alkyl aryl cross-couplings in water at rt
The nature of the zinc also plays an important role in the overall reactivity of this aqueous heterogeneous mixture. Insertion of Zn(0) into an alkyl iodide is influenced by size of the metal particles. Finely granulated zinc dust (up to 15 μm), as well as slightly larger zinc powder (15–40 μm), can both be used in these alkyl–aryl couplings. Attempted insertion reactions with commercially available nanoparticle zinc metal were extremely rapid and hence, uncontrolled, leading to mainly protioquenched alkyl halide. These reactions should also be run in an inert atmosphere to prevent exposure to oxygen, which otherwise does negatively impact the effectiveness/rate of electron transfer that presumably is taking place on the surface of the metal.
One catalyst particularly well-suited to these aqueous reactions is the Johnson Matthey palladium chloride complex PdCl2(Amphos)2 [16] (Scheme 1). The p-dimethylaminophenyl-di-tert-butylphosphine array appears to be important for increased levels of conversion, as the parent phenyl-di-tert-butylphosphine analogue gives inferior results. The scope of the reaction has been assessed on a variety of coupling partners bearing functional groups such as aldehydes, ketones, esters, N-protected amides, and silyl ethers, all being unaffected. Representative examples are illustrated in Scheme 2.

Representative examples of Zn-mediated, Pd-catalyzed couplings in water at rt
Surprisingly, the critical role of insulating the reactive RZnX from water played by a surfactant in alkyl–aryl C–C bond constructions seems to be unnecessary when benzylic rather than alkyl halides are used as partners [17]. Organozinc species formed “on the fly” in this case appear to rapidly participate in transmetalation in neat water at rates greater than that for protioquenching. A variety of valuable diarylmethanes can be made using functionalized benzylic chlorides or bromides together with mainly activated aryl iodides and bromides. Here again, zinc metal presumably inserts preferentially into Csp3–halogen bonds, whereas in situ-generated Pd(0) prefers an sp2 carbon–halogen bond. More easily handled benzylic chlorides (primary or secondary) are routinely used as coupling partners in this reaction. Zinc dust is an effective source of zinc for benzylic chlorides, whereas zinc powder is preferred for benzylic bromides (Scheme 3) [17].

Zn-mediated, Pd-catalyzed benzyl–aryl couplings in water at rt
An important feature of both described alkyl–aryl and benzyl–aryl couplings is that they are conducted at room temperature. Energy saved in avoiding both cooling and heating contributes to the overall “greening” of these procedures. Perhaps more noteworthy is that reactions under ambient conditions lead to fewer side reactions and hence, improved impurity profiles compared to reactions run at higher temperatures, thus simplifying purification. These reactions are remarkably simple to run: add the reactants to the reaction vessel containing either surfactant/water, or just water, then stir.
3 Aromatic C–H Bond Activation Using Cationic Palladium
In comparison with other cross-coupling transformations, transition metal-catalyzed aryl C–H bond activation reactions provide an especially direct and atom economical approach to functionalizing aromatic rings [18, 19]. In order to control regioselectivity and enhance reactivity of otherwise strong C–H bonds in arenes, various ortho-directing groups have been employed, with anilide-derivatives among the most convenient and broadly applicable substrates. Despite their strategic synthetic appeal, one major drawback has been the typically harsh conditions necessary to achieve activation of relatively inert aryl C–H bonds. Combinations of high temperatures, strong acids as solvent, and stoichiometric metal additives are frequently required. Recently, we have developed protocols that rely on highly reactive cationic palladium species to catalyze C–H arylations of aryl ureas with aryl iodides or boronic acids under much milder and greener conditions [20].
4 Aryl Iodides
Readily available aryl iodides have been commonly used as arylating agents. Extensive pioneering work, where halides serve as partners in direct C–H arylation of anilide derivatives, has been reported by the Sanford [21] and Daugulis [22, 23] groups. To realize products of couplings for this transformation, however, temperatures at or around 130 °C are common, along with strong protic acid such as TFA as solvent. Moreover, under such conditions, substrates are oftentimes prone to double ortho-arylation [23].
A gentler, more easily controlled, yet still direct arylation was envisioned, noting that the critical “C–H activation” step involves electrophilic attack of the metal onto the aryl nucleus via a palladacycle (Scheme 4). Key factors in determining the ease with which this intermediate is formed would clearly include the nature of the palladium catalyst itself as well as that of the directing group. The rate determining step apparently involves loss of the aryl hydrogen, as shown by a primary kinetic isotope effect [24]. For related mechanistic studies see: [25, 26]. A strongly cationic, and thus highly Lewis acidic palladium species should be especially reactive towards electrophilic H-atom substitution and have a particularly strong affinity for common anilide-type directing groups [27–29]. Careful selection of the specific ortho-directing anilide derivative would be expected to maximize this effect. Thus, tuning the cationicity of the palladium catalyst with corresponding adjustment of the chelating residue should minimize the extent of heating required, and allow C–H activation to proceed under milder conditions. It was further hoped that the high molar concentrations associated with normal micellar catalysis in water at ambient temperatures would enhance the ease and attractiveness of this reaction.

Formation of palladacycle by cationic palladium, followed by arylation
One method for increasing the Lewis acidity of palladium complexes involves the addition of acids with bulky or non-coordinating counter ions that favor formation of cationic palladium(II) salts (Scheme 5) [30]. Using a 2 wt% aqueous solution of PTS as the micelle-forming amphiphile, we investigated a variety of Pd(0) and Pd(II) sources as catalysts for ortho-directed C–H arylations with 4-iodoanisole as a model substrate. Inexpensive Pd(OAc)2 was found to be particularly active when used in conjunction with excess HBF4. Although the pre-formed, BF4-containing cationic palladium catalyst, [Pd(MeCN)4](BF4)2 is commercially available [31], it was ineffective for arylation with iodides; the presence of acetonitrile was found to severely suppress this reaction. As in the case of Daugulis’ chemistry, the presence of a silver(I) salt, such as AgOAc, was also found to be essential for reactivity, although its precise role is still unclear; mechanistic work is ongoing. With the goal of developing a coupling reaction that requires no external heating, the optimized catalyst system was screened against a number of anilide derivatives at room temperature. Only an N,N-dimethylurea-based substrate (Scheme 6) was found to react efficiently at room temperature to give the desired product. While a number of commercially available surfactants were effective, the highest yields were realized using the simple amphiphile Brij 35 [32].

Generation of cationic palladium species

Room temperature C–H arylations of aryl ureas with aryl iodides
These room temperature, organic solvent-free conditions were found to be general for couplings between a variety of aryl ureas and aryl iodides, done in air, yielding the desired ortho-arylated products in good-to-excellent yields (Scheme 6). Importantly, under such mild conditions, monoarylated products could be obtained for substrate types that previously led to uncontrolled double arylation [22], such as a simple unsubstituted phenyl urea. As has been previously observed [22, 23], contrary to the normal aryl halide cross-coupling reactivity profile, electron-rich aryl iodides were typically more reactive than their electron-deficient counterparts. Especially challenging substrates, including those bearing strongly electron-withdrawing groups and ortho-substituted iodides are currently still outside the scope of this methodology.
5 Aryl Boronic Acids
The palladium-catalyzed Suzuki–Miyaura cross-coupling of aryl halides and boronic acids has emerged as the method of choice for the synthesis of biaryls [33]. The ubiquity of biaryl targets, both in nature and of unnatural origin [34], has led to dramatic increases in commercial availability of aryl boronic acids and their derivatives. These stable organometallics have emerged as practical substrates for other arylation reactions as well. Although several examples have appeared using aryl boronic acids in ortho-directed C–H activation reactions with anilide derivatives, like most C–H activation protocols, these transformations generally require harsh conditions [35–38].
Typical Suzuki–Miyaura reactions employ a base to activate an organoboronic acid via ate complex formation, which more readily transmetalates the aryl group to palladium prior to product-forming reductive elimination [1]. In the presence of highly active cationic palladium catalysts, however, boronic acids alone can participate in aryl transfer, thereby completing the catalytic cycle without prior boron activation by base [29]. Thus, by virtue of both their inherent tendency toward palladacycle formation and their especially potent reactivity with aryl boron species, cationic palladium catalysts are expected to be particularly well-suited to C–H activation/cross-coupling with aryl boronic acids.
Unfortunately, however, conditions leading to the desired biaryl products from aryl ureas and arylboronic acids could not be found using a surfactant/water combination. Despite this unexpected setback, a simple switch to ethyl acetate (considered a green organic solvent due to its ready hydrolysis to innocuous components) gave very efficient couplings under some of the mildest conditions yet reported for ortho-directed C–H arylations (Scheme 7). A broad range of substrates can be utilized. Unlike C–H activation with aryl iodides, it was found that the commercially available cationic complex [Pd(MeCN)4](BF4)2 is effective in catalyzing the reaction with boronic acids, eliminating the need for addition of a strong external acid. Furthermore, in the presence of benzoquinone (BQ), silver additives are unnecessary. Thus, notwithstanding the need for an organic solvent, the conditions developed for this transformation are very mild, and among the greenest yet reported for C–H activation: no external acid, no stoichiometric metal oxidant, and no investment of energy beyond room temperature.

Room temperature C–H arylations of aryl ureas with arylboronic acids
Substrate scope for this reaction is also quite broad, allowing for the synthesis of biaryls not yet accessible in high yields through the aryl iodide route. These include both electron-poor aryl ureas, and educts possessing a wider range of functionality. Particularly noteworthy is the finding that an ortho-substituted aryl group can be introduced (Scheme 8). Double ortho-arylation is also generally well suppressed, with a high degree of selectivity for mono-arylated products even in cases with minimal substitution on the aryl ureas.

Representative examples of C–H arylations of aryl ureas with arylboronic acids
6 Summary and Conclusions
There are many incentives for enhancing the attractiveness of transition metal-catalyzed cross-coupling reactions. An important consideration today in developing new processes is that they be safer, easier, and cleaner from an environmental perspective. In this regard, we described herein C–C bond-forming reactions that are unusual; that rely on an in situ activation of alkyl carbon–halogen or aromatic carbon–hydrogen bonds. Thus, separate, discrete steps commonly used to generate reactive organometallic intermediates or reagents can be obviated, offering new “green” chemistry that is benign by design as an alternative to traditional approaches in organic media.
References
Negishi E (ed) (2002) Handbook of organopalladium chemistry for organic synthesis. Wiley, New York
Ojima I (ed) (2000) Catalytic asymmetric synthesis. Wiley, New York
Lipshutz BH, Yamamoto Y (eds) (2008) Chem Rev 108:2793
Mauger CC, Mignani GA (2006) Aldrichim Acta 39:17
Díez-González S, Nolan SP (2007) Top Organomet Chem 21:47
Sheldon R, Arrends IWCE, Hanefeld U (2007) Green chemistry and catalysis. Wiley-VCH, Weinheim, Germany
Anastas PT, Warner JC (1998) Green chemistry: theory and practice. Oxford University Press, New York
Corriu RJP, Masse JP (1972) J Chem Soc, Chem Commun 14
Tamao K, Sumitani K, Kumada M (1972) J Am Chem Soc 94:4374
Czaplik WM, Mayer M, von Wangelin AJ (2009) Angew Chem Int Ed 48:607
Lipshutz BH, Ghorai S (2008) Aldrichim Acta 41:59
Krasovskiy A, Duplais C, Lipshutz BH (2009) J Am Chem Soc 131:15592
Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB (2005) Angew Chem Int Ed 44:3275
Petrier C, Luche JL (1985) J Org Chem 50:910
Einhorn C, Luche JL (1987) J Organomet Chem 322:177
Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Rider PJ (2006) PdCl2(Amphos)2: CAS no. 887919-35-9. Org Lett 8:1787
Duplais C, Krasovskiy A, Wattenberg A, Lipshutz BH (2010) Chem Commun 46:562
Daugulis O, Do H-Q, Shabashov D (2009) Acc Chem Res 42:1074
Chen X, Engle KM, Wang D-H, Yu J-Q (2009) Angew Chem Int Ed 48:5094
Nishikata T, Abela AR, Lipshutz BH (2010) Angew Chem Int Ed 49:781
Kalyani D, Deprez NR, Desai LV, Sanford MS (2005) J Am Chem Soc 127:7330
Shabashov D, Daugulis O (2007) J Org Chem 72:7720
Daugulis O, Zaitsev VG (2005) Angew Chem Int Ed 44:4046
Xiao B, Fu Y, Xu J, Gong T-J, Dai J-J, Yi J, Liu L (2010) J Am Chem Soc 132:468
Balcells D, Clot E, Eisenstein O (2010) Chem Rev 110:749
Sehnal P, Taylor RJK, Fairlamb IJS (2010) Chem Rev 110:824
Lebrasseur N, Larrosa I (2008) J Am Chem Soc 130:2926
Houlden CE, Hutchby M, Bailey CD, Ford JG, Tyler SNG, Gagné MR, Lloy-Jones GC, Booker-Milburn KI (2009) Angew Chem Int Ed 48:1830
Nishikata T, Yamamoto Y, Miyaura N (2003) Angew Chem Int Ed 42:2768
Yamamoto A (1995) J Organomet Chem 500:337
[Pd(MeCN)4](BF4)2: CAS no. 21797-13-7
Brij 35 (polyoxoethylene (23) lauryl ether): CAS no. 9006-65-9
Miyaura N (2008) Bull Chem Soc Jpn 81:1535
Bringmann G, Walter R, Weirich R (1990) Angew Chem Int Ed 29:977
Chu J-H, Tsai S-L, Wu M-J (2009) Synthesis 3757
Wang D-H, Mei T-S, Yu J-Q (2008) J Am Chem Soc 130:17676
Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu J-Q (2007) J Am Chem Soc 129:3510
Chen X, Goodhue CE, Yu J-Q (2006) J Am Chem Soc 128:12634
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lipshutz, B.H., Abela, A.R., Bošković, Ž.V. et al. “Greening Up” Cross-Coupling Chemistry. Top Catal 53, 985–990 (2010). https://doi.org/10.1007/s11244-010-9537-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-010-9537-1