
Abstract
A solid Brønsted acid of amorphous carbon bearing SO3H, COOH and phenolic OH groups has been studied as a catalyst for biodiesel production. The carbon material functions as a stable and efficient catalyst for the formation of biodiesel from oleic acid at 353 K; the catalytic performance is 70–80% that of sulfuric acid. The carbon material also exhibits remarkable catalytic performance for transesterification of triolein at 403 K, maintaining high catalytic activity even in the presence of water. These results suggest that this catalyst can directly convert crude vegetable oils composed of triglycerides, free higher fatty acids and water into biodiesel with minimal energy consumption.
Similar content being viewed by others

1 Introduction
Recently, advances in production technology and changes in the political climate have increased the availability and awareness of biodiesel, an alternative to petroleum-derived diesel fuel with much lower net-sum CO2 emissions due to the absorption of CO2 by plants used to produce the fuel. Biodiesel is, in a broad sense, composed of higher fatty acid derivatives, such as fatty acid esters made from triglycerides (esters of higher fatty acids and glycerol) or higher fatty acids in plant materials or animal fat. Plants convert solar-energy into plant materials, such as cellulose, starch, saccharides, lignin, higher fatty acids and triglycerides, while absorbing CO2. The conversion efficiency from solar-energy into plant material reaches a maximum of ca. 3%. Triglycerides and higher fatty acids contained in plant seeds cannot be directly combusted in modern diesel engines; therefore, these are converted into biodiesel using chemical processes that consume energy. Combustion of biodiesel in diesel engines results in the production of energy and CO2, which is then absorbed through plant growth. The total energy invested for the cultivation of plants that bear seeds containing starting materials, the harvest and transport of the seeds, and conversion of the seeds into biodiesel is lower than the energy of the biodiesel produced (life cycle assessment (LCA) > 1); therefore, biodiesel production and its use is genuine solar-energy conversion, and the biodiesel is renewable clean energy without CO2 emissions. On the other hand, in the case of LCA < 1, the “biodiesel” is not renewable energy and is not carbon neutral. Therefore, for the production of environmentally benign biodiesel, it is essential to minimize energy consumption for all processes, especially the production of plant seeds and chemical processes used to convert the plant seed components into biodiesel [1, 2].
Crude vegetable oils obtained by compressing plant seeds or fruits, such as soybeans, rapeseed and palm fruit, are starting materials for large-scale biodiesel production. The crude vegetable oils consist of considerably free higher fatty acids and water, in addition to triglycerides. Many familiar vegetable cooking oils that consist of triglycerides are obtained by the removal of higher fatty acids and water from the crude oils, followed by purification. While the conversion of waste vegetable cooking oils into biodiesel is an effective method for recycling of vegetable cooking oils, the amount of biodiesel produced from the starting materials is too small to be practically useful for the fueling of all diesel engine vehicles. Therefore, vegetable cooking oils should be distinguished from crude vegetable oils for the large-scale production of biodiesel. Direct conversion of crude vegetable oils into biodiesel is favorable for the large scale and environmentally benign production of biodiesel. However, it is difficult for any base catalyst to produce biodiesel from crude vegetable oils, because water in the starting materials largely decreases base catalysis, and neutralization of basic active sites in the catalysts by higher fatty acid prevents biodiesel production by transesterification. One process for biodiesel production from crude vegetable oil is schematically shown Fig. 1, which illustrates biodiesel production by the esterification of higher fatty acids and the transesterification of triglycerides. The most popular method is acid catalyzed esterification, followed by base-catalyzed transesterification. In this process, the use of homogeneous or soluble acid and base catalysts, as in conventional biodiesel production, results in a large environmental load. Although homogeneous acid catalysts such as H2SO4 exhibit high catalytic performance for the esterification of higher fatty acids, they require special processing in the form of neutralization, in addition to costly and inefficient catalyst separation. The use of soluble base catalysts such as KOH for transesterification of triglycerides in the product obtained by esterification also leads to large energy consumption.

Biodiesel production from crude vegetable oils by esterification–transesterification process
A heterogeneous catalyst that functions as a stable and efficient catalyst for both reactions would be applicable to the environmentally benign production of biodiesel from crude vegetable oils; such a catalyst should be readily separated from the product and be reusable for repeated reaction with minimum energy consumption and without waste. Solid acid catalysts are promising candidates for this purpose, because acids can catalyze both esterification and transesterification. However, the transesterification of triglycerides by acid catalysts usually requires high reaction temperatures of more than ca. 423 K [1]. In this study, the esterification of oleic acid and transesterification of triolein over amorphous carbon bearing SO3H, COOH and phenolic OH groups were examined for potential application to environmentally benign biodiesel production. The carbon material referred to as sugar catalyst [2–5] is new type of amorphous carbon-based solid acid catalyst composed of nano-graphene bearing SO3H groups [6]. This carbon material, which exhibits remarkable catalytic performance for biodiesel production by esterification of higher fatty acids [3, 4], was designed to be a highly active, stable and reusable solid acid as an environmentally benign replacement for H2SO4 [6].
2 Experimental
2.1 Preparation of Carbon Material Bearing SO3H Groups
The carbon material with SO3H groups was prepared from microcrystalline cellulose powder (Avicel, Merck; particle size, 20–100 μm; crystallinity, 80%; degree of polymerization, 200–300). The starting material (20 g) was heated for 5 h at 723 K under N2 flow to produce a black solid, which was then ground to a powder. The powder (7 g) was then boiled in 150 cm3 of fuming sulfuric acid (15 wt% SO3) at 353 K under N2. After heating for 10 h and then cooling to room temperature, the suspension was filtered to yield a black precipitate, which was washed repeatedly with hot distilled water (>353 K, 1,000 cm3) until impurities such as sulfate ions were no longer detected in the wash water. As only a small amount of the excess fuming sulfuric acid is consumed in the reaction, the sulfuric acid recovered after filtration of the powder can be reused for repeated sulfonation of the carbon material. The densities of the functional groups in the carbon material are mainly dominated by the degree of carbonization in the carbon material before sulfonation [7], and the degree of carbonization can be controlled with good reproducibility; therefore, a reproducible carbon material can be easily prepared.
2.2 Characterization
Structural information for the prepared carbon material was obtained by scanning electron microscopy (SEM; S-5200, Hitachi), powder X-ray diffraction (XRD; Rint 2100 diffractometer, Rigaku), Raman spectroscopy (NRS-2100, Jasco), Fourier transform infrared spectroscopy (FTIR; FT/IR-6100, Jasco), 13C cross-polarization (CP) magic angle spinning (MAS) nuclear magnetic resonance (CP/MAS NMR; Bruker ASX-200), 31P MAS NMR, and gas adsorption analysis. FTIR spectra of the carbon material were obtained using KBr pellets containing the prepared carbon powder. 13C CP/MAS NMR spectra were measured at room temperature with a Larmor frequency of 50.3 MHz. A Bruker MAS probe head was used with a 7 mm zirconia rotor and a sample spinning rate of 4.0 kHz. The frequency of the spectra is expressed with respect to neat tetramethylsilane. Experimentally, glycine was used as a second reference material, with the carbonyl signal set at 176.48 ppm. The acid strength of the carbon material was examined using color-producing reagents and 31P MAS NMR measurements with trimethylphosphine oxide (TMPO) as a probe molecule [8]. The Brunauer–Emmett–Teller (BET) surface areas and water vapor adsorption isotherms for the samples were measured using a Nova-4200e (Quantachrome) and an Autosorb MP/VP (Quantachrome), respectively.
The amount of functional groups bonded to the carbon material was estimated by elemental analysis (vario MICRO cube, Elementar Inc.) and cation-exchange analysis. According to X-ray photoelectron spectroscopy (XPS) analysis, all sulfur in the carbon material, which possesses SO3H, COOH, and phenolic OH, is expected to be confined to SO3H groups [7]. The densities of SO3H groups were thus estimated based on the sulfur content determined from sample compositions obtained by elemental analysis. The total SO3H + COOH and SO3H + COOH + OH contents were estimated from the exchange of Na+ in aqueous NaCl and NaOH solutions, respectively, to afford the proportions of each functional group [9].
2.3 Esterification of Free Higher Fatty Acids and Transesterification of Triolein
Esterification of oleic acid and transesterification of triolein were carried out in a sealed Pyrex reactor containing the catalyst, methanol and oleic acid or triolein at 368 and 403 K, respectively. After the reaction, the reaction solution was analyzed by high performance liquid chromatography, gas chromatography (LC-2000, Jasco) and gas chromatograph mass spectrometry (GC17A/GSMS-QP5050).
3 Results and Discussion
3.1 Structure of the Carbon Material
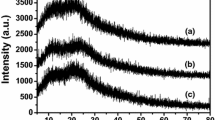
The particle size and surface area of the black powder obtained by sulfonation of partially carbonized cellulose were estimated by SEM and BET measurements to be 10–40 μm and 2 m2 g−1, respectively. The XRD pattern for the carbon exhibited two broad but weak diffraction peaks at 2θ angles of 10–30° and 35–50°, which is attributable to amorphous carbon composed of aromatic carbon sheets oriented in a considerably random fashion [7, 10]. The Raman spectrum for the sample showed that the intensity ratio of the D band (1,350 cm−1, A1g D breathing mode) to the G band (1,580 cm−1, E2g G mode) for the carbon material is 0.81, which indicated the average graphene size in the amorphous carbon was ca. 1 nm [9, 11]. The vibration bands at 1,040 (SO3 − stretching) and 1,377 cm−1 (O=S=O stretching in SO3H) in the FTIR spectrum of the carbon material confirmed that the resulting material has SO3H groups [9]. The broad band at 2,300–2,700 cm−1 was assigned to an overtone (Fermi resonance) of the bending mode of –OH·····O= linked by a strong hydrogen bond, as seen in strong liquid Brønsted acids such as CF3SO3H [9, 12], which suggests that some SO3H groups are in close proximity to each other. Chemical shifts observed at 130, 155, and 180 ppm in the 13C CP/MAS NMR spectrum of the sample are attributable to polycyclic aromatic carbon atoms, phenolic OH, and COOH groups, respectively [9]. Elemental analysis and cation-exchange experiments revealed that the sample composition is CH0.622O0.540S0.048 and that the amounts of SO3H, COOH, and phenolic OH groups bonded to the graphene are 1.9, 0.4 and 2.0 mmol g−1, respectively. These results indicate that the minimum unit in the material is a nano-graphene sheet (ca. 1 nm) composed of 10–20 6-membered rings. The graphene possesses a high density of functional groups such as SO3H, COOH and phenolic OH groups. If the carbon material is composed of uniform functionalized graphene sheets, then each graphene sheet is expected to bind SO3H, COOH and 3 phenolic OHs [9]. This is distinct from conventional solid acids with single functional groups. From the broad carbon (002) diffraction at 2θ angles of 10–30° in the XRD pattern, it is expected that several graphene sheets bearing functional groups are accumulated to form a domain. These domains are aggregated, constituting a catalyst particle (typically 10–40 μm diameter) similar to amorphous carbon, as shown in Fig. 2. The Hammet function of SO3H groups in conventional sulfonated polystyrene-based resins is H0 > −3. However, the carbon material has SO3H groups with H0 = −8 to −11, which is comparable to that of concentrated H2SO4 [9], although the reason for the strong acidity remains to be clarified. One possible explanation for the strong acidity of the SO3H groups is SO3H groups linked by hydrogen bonds. Some SO3H groups in the carbon materials are linked by strong hydrogen bonds, which can result in strong acidity due to mutual electron-withdrawal.

Incorporation of neutral hydrophilic molecules into the bulk of the carbon material
It should be noted that SO3H-bearing amorphous carbon incorporates a large amount of hydrophilic molecules, including water, into the bulk. The Brunauer–Emmett–Teller (BET) surface area of the carbon material after dehydration is only 2 m2 g−1, nevertheless, it has high catalytic activity for various acid-catalyzed reactions [3–7, 9]. Figure 3 shows a H2O vapor adsorption–desorption isotherm for the carbon material at 298 K. At 1.5 kPa, which is much less than the saturated water vapor pressure (ca. 3 kPa), the amount of adsorbed H2O exceeds 0.01 mol g−1. This indicates that the carbon material can incorporate a large amount of water into the carbon bulk, comparable to that of ion-exchangeable resins. XRD measurements revealed that the carbon interlayer spaces do not expand after water incorporation. Although this indicates that incorporated water molecules are not intercalated into the interlayer spaces between carbon sheets, the apparent volume of carbon particles after water incorporation increases by 1.2–1.5 times that of the carbon material prior to water incorporation. These results suggest that a large amount of water molecules are incorporated into the vacancies among the domains composed of grapheme, as shown in Fig. 2. It was also observed that other hydrophilic molecules, including large molecules such as butanol and cellohexaose (a polysaccharide of 6 glucose monomers linked by β-1,4 glycosidic bonds), are also incorporated into the carbon bulk, [13] which provides good access to the SO3H groups in the carbon material by hydrophilic reactants in solution and gives rise to high catalytic performance, despite the low surface area [7, 9, 13].

H2O vapor adsorption–desorption isotherm of carbon material at 298 K
3.2 Esterification and Transesterification Using Amorphous Carbon Bearing SO3H, COOH and Phenolic OH Groups
Table 1 shows the results for the carbon catalyst reuse experiment for esterification of oleic acid and methanol at 368 K. The methyl oleate yield reaches ca. 90% within 2 h in the presence of the carbon material. The carbon material exhibits much higher catalytic performance for the reaction than other tested conventional solid Brønsted acid catalysts, such as niobic acid (Nb2O5·nH2O), Amberlyst-15 (sulfonated polystyrene) and Nafion NR50 (perfluoro-sulfonated ionomer) [3], and is 70–80% that of sulfuric acid under the same reaction conditions. After the reaction for 4 h, the carbon catalyst was readily precipitated in the reaction vessel, and the precipitated catalyst was repeatedly reused for subsequent reactions after washing with water and drying at 403 K. No decrease in activity was observed even after 10 reuses, as long as the recovered catalyst was washed with water before the subsequent reaction. However, when the recovered catalyst was washed with methanol, the catalytic activity decreased with each catalyst reuse, reaching a plateau at 75–80% yield, as shown in Table 2. This can be attributed to the formation of methyl sulfonate from methanol and a part of the sulfonic acid groups bonded to the carbon material. The activity of the catalyst after 4 reuses was recovered by washing with water, as shown in Table 2.
The carbon material also shows high catalytic performance for the transesterification of triolein, as summarized in Tables 3 and 4. While SAC-13 [14] has much higher TON for the transesterification than Amberlyst-15 and Nafion NR50, Amberlyst-15 shows the largest methyl oleate yield because of a high density of SO3H groups in Table 3. The carbon material exhibits much higher methyl oleate yield and TON than these tested conventional solid acids. The formation of oleic acid and glycerol monooleate was observed for entry A of Table 4. The oleic acid formation can be attributed to the hydrolysis of triolein by water in the carbon material because the catalyst able to absorb a large amount of water as shown in Fig. 3 was used for the reaction without dehydration. The formation of glycerol monooleate is not favorable for high grade biodiesel production, because there is very little difference in the boiling points of the higher fatty acid-monoglyceride and the higher fatty acid–methanol ester, which results in separation difficulties. However, the addition of a small amount of water to the reaction system (B) decreases the formation of oleic acid and glycerol monooleate. Further addition of water to the reaction system (C) suppresses the formation of glycerol monooleate, although the amount of oleic acid formation in C is larger than that of B. The increase in oleic acid is due to the hydrolysis of triolein by the addition of water. Water in the reaction system is liable to poison base catalysts for conventional biodiesel production by transesterification; therefore, it is necessary to achieve efficient base-catalyzed triglyceride transesterification to dehydrate the triglycerides. On the other hand, reaction with the carbon catalyst does not significantly dependent on the amount of water in the reaction system, as shown in Table 4; therefore, transesterification of the triglyceride proceeds on the carbon material without sufficient dehydration, which is a costly and energy inefficient process, of the oil phase (a mixture of biodiesel, triglycerides and water) obtained through acid-catalyzed esterification of higher fatty acids in crude vegetable oils. In addition, transesterification by the carbon catalyst is efficient, with suppression of free higher fatty acid and monoglyceride formation in the presence of an adequate amount of water. Table 5 shows the results for the carbon catalyst reuse experiment for transesterification of triolein and methanol at 403 K under optimal reaction conditions. After the reaction for 5 h, the carbon catalyst was readily precipitated in the reaction vessel, and the precipitated catalyst was repeatedly reused for subsequent reactions after washing with water and drying at 403 K. No decrease in activity was observed even after 4 reuses, meaning that the carbon material function as a stable catalyst even under such harsh condition.
Such high catalytic performance of this carbon material, which is not observed for other solid acid catalysts, cannot be adequately explained simply by the high density of strongly acidic SO3H groups and good access of reactants to the SO3H groups in the carbon material by incorporation. Perfluoro-sulfonated ionomers such as Nafion, which can also incorporate a large amount of hydrophilic molecules into the bulk structures similar to the carbon material, and with a high density of more acidic SO3H groups (−13 < H0 < −11), have moderate activity for the esterification of free higher fatty acids and transesterification of triglycerides [2, 3, 8]. One possible explanation for the high performance of the carbon material bearing SO3H, COOH and phenolic OH groups may be the strong affinity between the hydrophilic parts of the reactants and the almost neutral OH groups bonded to graphene. The OH groups in the carbon material can adsorb cellulose by hydrogen bonding between the OH groups in the carbon material and cellulose, so that the carbon material can efficiently hydrolyze cellulose into glucose as well as sulfuric acid [9, 13]. In contrast to the carbon material, conventional solid acids (inorganic oxide- and SO3H-bearing polymer-based solid acids) cannot hydrolyze cellulose at all [9, 13], because the former does not have effective acid sites to function in the presence of water, and hydrophobic substrate and acid sites in the latter do not have an affinity with hydrophilic cellulose. Both esterification and transesterification are reactions that involve alcohols, which are hydrophilic molecules. As a result, the incorporation of alcohols into highly hydrophilic carbon material may contribute to the high catalytic performance. In fact, it was confirmed that SO3H-bearing amorphous carbon without OH groups (SO3H: 4.9 mmol g−1) [6], prepared by heating naphthalene in conc. H2SO4, cannot absorb lager amount of alcohol than amorphous carbon with SO3H, COOH and phenolic OH groups (the absorbed amount of alcohol on the former is ca. 20% that of the latter) and has moderate activity for the reactions as well as Amberlyst-15. Decrease in the formation of oleic acid and glycerol monooleate over the carbon material in the presence of adequate amount of water is not also explained by conventional acid and base catalyses, including homogeneous acid and base catalysts, suggesting that such behaviors are not due only to SO3H groups bonded to graphene: a collaboration among water and the functional groups results in transesterification peculiar to the carbon material. Correlation of the carbon structure with the acid-catalyzed reactions has not yet been clarified, but further detailed investigations are in progress.
4 Conclusions
Amorphous carbon-bearing SO3H, COOH and phenolic OH groups functions as an efficient heterogeneous catalyst for biodiesel production by esterification of oleic acid and transesterification of triolein. Transesterification in the presence of an adequate amount of water suppresses the formation of by-products. These results suggest that the carbon catalyst, a heterogeneous catalyst that is easily separated from the products, is available for the environmentally benign production of biodiesel from crude vegetable oils.
References
Serio MD, Tesser R, Pengmei L, Santacesaria E (2008) Energy Fuels 22:207
Hara M (2009) ChemSusChem 2:129
Toda M, Takagaki A, Okamura M, Kondo JN, Hayashi S, Domen K, Hara M (2005) Nature 438:178
Zong M-H, Duan Z-Q, Lou W-Y, Smith TJ, Wu H (2007) Green Chem 9:434
Budarin VL, Clark JH, Luque R, Macquarrie DJ (2007) Chem Commun 6:634
Hara M, Yoshida T, Takagaki A, Takata T, Kondo JN, Domen K, Hayashi S (2004) Angewe Chem Int Ed 43:2955
Okamura M, Takagaki A, Toda M, Kondo JN, Tatsumi T, Domen K, Hara M, Hayashi S (2006) Chem Mater 18:3039
Nakajima K, Hara M (2007) J Am Ceram Soc 90:3725
Suganuma S, Nakajima K, Kitano M, Yamaguchi D, Kato H, Hayashi S, Hara M (2008) J Am Chem Soc 130:12787
Tsubouchi N, Xu K, Ohtsuka Y (2003) Energy Fuels 17:1119
Ferrari AC, Robertson J (2000) Phys Rev B 61:14095
Buzzoni R, Bordiga S, Ricchiardi G, Spoto G, Zecchina A (1995) J Phys Chem 99:11937
Kitano M, Yamaguchi D, Suganuma S, Nakajima K, Kato H, Hayashi S, Hara M (2009) Langmuir 25:5068
López DE, Goodwin JG, Bruce David A (2007) J Catal 245:381
Acknowledgments
This work was supported by the New Energy and Industrial Technology Development Organization (NEDO, 04A32502), the Research and Development in a New Interdisciplinary Field Based on Nanotechnology and Materials Science programs of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, a Grant-in-Aid for Scientific Research (18206081) from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Environment (MOEN).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hara, M. Biodiesel Production by Amorphous Carbon Bearing SO3H, COOH and Phenolic OH Groups, a Solid Brønsted Acid Catalyst. Top Catal 53, 805–810 (2010). https://doi.org/10.1007/s11244-010-9458-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-010-9458-z