
Abstract
Three metal–organic complexes with two anthracene-based bulky backbone ligands 9,10-dioxo-9,10-dihydroanthracene-1,5-dicarboxylic acid (H2 L 1) and 2-phenylquinoline-4-carboxylic acid (HL 2), namely {[Mn(L 1)0.5(L 1)0.5(phen)(H2O)2](H2O)1.5}∞ (1), [Mn2(L 2)4(phen)2(H2O)2](H2O)2 (2) and [Cd3(L 2)2(pp)2Cl4]∞ (3) (phen = 1,10-phenanthroline, pp = 3-(2-pyridyl)pyrazole), have been synthesized and characterized. X-ray single-crystal diffraction indicates that H2 L 1 adopts a bis-monodentate bridging mode in complex 1, whereas HL 2 adopts monodentate and bidentate bridging modes in 2 and a tridentate bridging mode in 3. The chelating ligands phen and pp give rise to a 1D zigzag chain in complexes 1 and 3, and a binuclear structure in 2, by preventing the formation of higher-dimensional structures. Notably, the anions play an important role in the structure of complex 3. In addition, the magnetic and luminescent properties of these complexes were investigated.
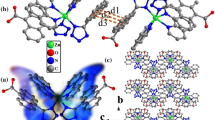
Graphical Abstract
Three metal–organic compounds, showing a 1D zigzag chain, a binuclear structure and a 1D zigzag chain, have been successfully constructed by using 9,10-dioxo-9,10-dihydroanthracene-1,5-dicarboxylic acid and 2-phenylquinoline-4-carboxylic acid, together with incorporating auxiliary bridging ligands 1,10-phenanthroline or 3-(2-pyridyl)pyrazole. The present results reveal that chelated ligands phen and pp play an important role in the formation of frameworks of 1–3, which prevent the formation of higher-dimensional structures. Moreover, intense luminescent emissions of 1–3 were attributed to the LLCT of corresponding ligand. And antiferromagnetic coupling interaction exists between the binuclear MnII ions in 2.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The design of metal–organic crystalline materials has attracted considerable attention due to their interesting architectures and applications in optics, magnetism, gas storage, ion exchange and catalysis [1, 2]. The structural and functional properties of such target materials vary significantly with the metal and ligand choice [1, 2]. In this respect, a variety of organic building blocks containing pyridyl and/or carboxylate functional groups have been intensely investigated [1–4], and the rational design of new types of organic ligands for constructing unusual coordination polymers is obviously not a trivial problem at this stage. Ligands with bulky backbones are important in the field of coordination chemistry, because of their steric hindrance effects [5, 6]. Very recently, increasing numbers of investigations have been focused on the self-assembly of such bulky backbone multidentate ligands with metals. This can lead to the formation of either discrete polynuclear complexes or low-dimensional coordination polymers, which display such useful properties as luminescence, and sensor ability. Additionally, auxiliary ligands [7], counteranions [8], as well as synthesis conditions [9], including the pH value of the reaction solution [10], temperature [11], molar ratio between reactants [12] and solvent system [13], can all play an important role in the formation of such complexes.
In our previous work, several anthracene-based bulky backbone ligands, namely acridine-9-carboxylic acid [14], anthracene-9-carboxylic acid [15], anthracene-9,10-dicarboxylic acid [15, 16] and anthracene-1,5-dicarboxylic acid [16], were used to construct a range of metal coordination compounds, showing interesting luminescent, magnetic and gas adsorption properties. By contrast, anthracene-based bulky backbone ligands, such as phenylquinoline- [17, 18] and anthraquinone-based carboxylic acids [19, 20], have not yet been widely studied. Therefore, we have selected two anthracene-based bulky backbone ligands, 9,10-dioxo-9,10-dihydroanthracene-1,5-dicarboxylic acid (H2 L 1) and 2-phenylquinoline-4-carboxylic acid (HL 2) (Fig. 1), together with 1,10-phenanthroline (phen) or 3-(2-pyridyl)pyrazole (pp) as auxiliary ligands, to construct three MnII and CdII coordination compounds, namely {[Mn(L 1)0.5(L 1)0.5(phen)(H2O)2](H2O)1.5}∞ (1), [Mn2(L 2)4(phen)2(H2O)2](H2O)2 (2) and [Cd3(L 2)2(pp)2Cl4]∞ (3) (phen = 1,10-phenanthroline, pp = 3-(2-pyridyl)pyrazole). To facilitate the generation and crystallization of these complexes, the hydrothermal synthetic method has been employed [21]. The magnetic and luminescent properties of these complexes have been investigated.

The ligands used in this work
Experimental
Materials and methods
9,10-Dioxo-9,10-dihydroanthracene-1,5-dicarboxylic acid (H2 L 1, Scheme 1) was synthesized according to the literature procedure [22]. All other reagents and solvents for synthesis were commercially available and used as received or purified by standard methods prior to use. Elemental analyses (C, H, N) were obtained on a Perkin–Elmer 240C analyzer. IR spectra were recorded on a Varian 800 (Scimitar Series) FT-IR spectrometer with KBr pellets. Emission spectra in the solid state were taken on a Cary Eclips fluorescence spectrophotometer at room temperature. Magnetic measurements were carried out with a quantum design MPMS-XL-7 magnetometer.

Synthetic route for H2 L 1
Preparation of complex 1
A mixture of H2 L 1 (148 mg, 0.5 mmol), phen (99 mg, 1.2 mmol) and MnCl2·2H2O (324 mg, 2 mmol) was dissolved in water (15 mL). The resultant solution was sealed in a Teflon-lined stainless autoclave and heated to 160 °C. After keeping in these conditions for 3 days, the reaction vessel was allowed to cool. Yellow single crystals suitable for X-ray analysis were obtained after cooling to room temperature. Yield: ~40 % based on H2 L 1. Calcd. (%) for C56H42N4O19Mn2: C, 56.8; H, 3.6, N, 4.7; found (%): C, 56.3; H, 3.9, N, 4.3. IR (KBr pellet, cm−1): 3,374 s(br), 3,075 w, 1,689 s, 1,567 s, 1,517 s, 1,466 m, 1,438 w, 1,407 s, 1,326 s, 1,275 s, 1,232 w, 1,167 w, 1,137 w, 1,094 w, 1,008 w, 865 w, 844 w, 827 m, 807 w, 783 w, 770 m, 727 m, 711 s, 686 m, 614 w, 421 w.
Preparation of complex 2
An ethanol solution (10 mL) of HL 2 (13 mg, 0.05 mmol), phen (9 mg, 0.05 mmol) and excess 2,6-dimethylpyridine (ca. 0.05 mL for adjusting the pH value) was carefully layered on top of an aqueous solution (15 mL) of Mn(ClO4)2·6H2O (36 mg, 0.1 mmol) in a test tube. Yellow single crystals suitable for X-ray analysis appeared at the boundary between the layers after ca. 1 month at room temperature. Yield: ~30 % based on HL 2. Calcd. (%) for C88H64N8O12Mn2: C, 68.8, H, 4.2, N, 7.3; found (%): C, 69.1, H, 4.5, N, 7.4. IR (KBr pellet, cm−1): 3,464 s(br), 1,692 s, 1,573 s, 1,548 m, 1,517 s, 1,458 m, 1,427 s, 1,380 s, 1,373 s, 1,319 m, 1,234 m, 1,140 m, 1,025 m, 914 w, 865 w, 813 m, 769 s, 726 m, 651 m, 567 w, 514 w, 411 w.
Preparation of complex 3
The same procedure as that for 2 was used for this complex except that Mn(ClO4)2·6H2O and phen were replaced by CdCl2·2.5H2O and pp, respectively. Light yellow single crystals suitable for X-ray analysis were obtained after cooling to room temperature. Yield: ~20 % based on HL 2. Calcd. (%) for C48H34N8O4Cl4Cd3: C, 45.4, H, 2.7, N, 8.8; found (%): C, 45.6, H, 2.5, N, 8.6. IR (KBr pellet, cm−1): 3,473 s(br), 3,126 m, 1,641 w, 1,601 m, 1,570 s, 1,544 m, 1,463 s, 1,432 s, 1,365 m, 1,313 m, 1,237 w, 1,153 m, 1,095 m, 1,068 w, 1,023 w, 970 w, 879 w, 848 w, 808 w, 783 m, 764 s, 694 m, 637 m, 590 m, 515 w, 411 w.
X-ray powder diffraction studies
The X-ray powder diffraction patterns (XRPD) of complexes 1–3 were recorded on a Rigaku D/Max-2,500 diffractometer, operated at 40 kV and 100 mA, using a Cu-target tube and graphite monochromator. The intensity data were recorded by continuous scan in 2θ/θ mode from 3° to 40° with a step size of 0.02° and a scan speed of 8° min−1.
X-ray crystallographic studies
X-ray single-crystal diffraction measurements for complexes 1–3 were carried out on a Bruker Smart 1000 CCD diffractometer equipped with a graphite crystal monochromator situated in the incident beam for data collection at 293(2) K. The determinations of unit cell parameters and data collections were performed with Mo-Kα radiation (λ = 0.71,073 Å), and unit cell dimensions were obtained with least-square refinements. The program SAINT [23] was used for integration of the diffraction profiles. Semi-empirical absorption corrections were applied using the SADABS program [24]. All the structures were solved by direct methods using the SHELXS program of the SHELXTL package and refined with SHELXL [25]. Metal atoms in each complex were located from the E-maps, and other non-hydrogen atoms were located in successive difference Fourier syntheses and refined with anisotropic thermal parameters on F 2. The hydrogen atoms were added theoretically, riding on the atoms concerned and refined with fixed thermal factors. Crystallographic data and experimental details for the structural analyses are summarized in Table 1. Selected bond distances and angles are listed in Table 2. CCDC nos 942732, 942731 and 942730 contain the supplementary crystallographic data for complexes 1–3, respectively. This material can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or E-mail: deposit@ccdc.cam.ac.uk.
Results and discussion
Synthesis and general characterization
In general, reducing the reaction speed may result in the slow growth of well-shaped larger single crystals suitable for X-ray diffraction [26]. With this in mind, the synthesis and isolation of complexes 2 and 3 were carried out through self-assembly reactions of Mn(ClO4)2·6H2O or CdCl2·2.5H2O with HL 2, together with phen or pp as chelating co-ligands, by using the slow diffusion method under mild conditions of ambient temperature and pressure. However, under the same conditions, the reaction of MnII and H2 L 1 gave only some precipitate or microcrystalline products. Hydrothermal synthesis can minimize the problems associated with ligand solubility as well as enhancing the reactivity of reactants, so well-shaped single crystals of 1 were achieved by the high-temperature hydrothermal synthesis. Complexes 1–3 are all air stable. The compositions of these new materials were validated by elemental analyses and IR spectra. The phase purities of the bulk samples were checked by XRPD (see Fig. 7).
Crystal structure of complex 1
Similar to previous reports [17, 27, 28], X-ray single-crystal diffraction analysis reveals that the asymmetric unit of 1 contains one MnII atom, two half L 1 ligands, one phen ligand, two coordinated water ligands and one and three half water molecules (Fig. 2). In other words, each MnII center is six coordinated by two carboxylate O atoms from two L 1 ligands, two N atoms from one phen ligand and two O atoms from two water ligands, forming a slightly distorted octahedral coordination geometry. Each L 1 ligand coordinates to two MnII atoms via its two carboxylate groups. Thus, the MnII centers are linked by L 1 ligands via a bis-monodentate bridging mode to result in 1D zigzag chains, which are prevented from forming higher-dimensional structures by the chelating phen ligands (Fig. 2a). The 1D zigzag chains are assembled to form a 3D supramolecular network by intermolecular C–H\( \cdots \)O hydrogen bonds [the C(3)\( \cdots \)O(1G) and C(11)\( \cdots \)O(4H) separations are 3.44(8) and 3.43(4) Å and the C(3)–H(3)\( \cdots \)O(1G) and C(11)–H(11)\( \cdots \)O(4H) angles are 155.00° and 162.01°, symmetry code G = 1 − x, −y, 1 − z; H = 1 − x, −y, −z] (Fig. 2b) [29, 30].

View of a the coordination environment of MnΙΙ ions in the zigzag chain of 1, b the 3D supramolecular network showing the intramolecular C–H\( \cdots \)O hydrogen bond interactions (partial H atoms and dissociated aqua molecules omitted for clarity)
Crystal structure of complex 2
When we used HL 2 instead of H2 L 1, a reported binuclear complex 2 was produced (Fig. 3) [31]. The MnII centers are linked by L 2 ligands via a bidentate bridging mode to form a binuclear structure, which is prevented from forming higher-dimensional structures by the chelating phen and L 2 ligands in a monodentate mode (Fig. 3a). The binuclear units are assembled to form a 3D supramolecular network by intermolecular O–H\( \cdots \)O hydrogen bonds [the O(2W)\( \cdots \)O(3C) separation is 3.18(1) Å, and the O(2W)–H(2WB)\( \cdots \)O(3C) angle is 169.67°, symmetry code C = 1 − x, −y, 1 − z], O–H\( \cdots \)N hydrogen bonds [the O(1W)\( \cdots \)N(1D) and O(2W)\( \cdots \)N(2E) distances are 2.87(1) and 3.05(1) Å, and the O(1W)–H(1WB)\( \cdots \)N(1D) and O(2W)–H(2WA)\( \cdots \)N(2E) angles are 175.11° and 162.08°, symmetry code D = −x, 0.5 + y, 1.5 − z; E = x, y, −1 + z] and also C–H\( \cdots \)O hydrogen bonds [the C(40)\( \cdots \)O(1WF) separation is 3.48(1) Å, and the C(40)–H(40)\( \cdots \)O(1WF) angle is 167.39°, symmetry code F = x, 0.5 − y, −0.5 + z] (Fig. 3b) [29, 30].

View of a the coordination environment of MnΙΙ ions in the dinuclear unit of 2, b the 3D supramolecular network showing the intramolecular O–H\( \cdots \)O, O–H\( \cdots \)N and C–H\( \cdots \)O hydrogen bond interactions (partial H atoms, partial ligand backbones and dissociated aqua molecules omitted for clarity)
Crystal structure of complex 3
Different from complex 2, the asymmetric unit of complex 3 is composed of one and a half CdII atoms, one L 2 ligand, one pp ligand and two Cl− ligands. The two CdII centers have different coordination spheres (see Fig. 4a; Table 2). Cd(1) is coordinated by two O atoms from two L 2 ligands and four Cl− ligands, giving a distorted octahedral geometry. Cd(2) is six coordinated by two O atoms from two L 2 ligands, two N atoms from one pp ligand and two Cl− ligands to form a distorted octahedral geometry. Thus, pairs of CdII centers are linked by L 2 ligands via a tridentate bridging mode and bridging Cl− ligands to result in a 1D zigzag chain (Fig. 4b). These 1D chains are assembled into a 2D supramolecular network by intermolecular C–H\( \cdots \)Cl hydrogen bonds [the C(15)\( \cdots \)Cl(2C) separation is 3.49(4) Å, and the O(15)–H(15A)\( \cdots \)Cl(2C) angle is 132.86°, symmetry code C = x, −y + 2, z − 1/2] [29, 30] and π \( \cdots \) π stacking interactions between pyridyl and benzene rings [the centroid–centroid and interplanar separations are 3.58(8) and 3.43(8) Å, respectively, with the dihedral angle being 7.3°] [32, 33] (Fig. 4c).

View of a the coordination environment of CdΙΙ ions in the dinuclear unit of 3, b the 2D supramolecular network showing the intramolecular C–H\( \cdots \)Cl hydrogen bond and π \( \cdots \) π stacking interactions (partial H atoms omitted for clarity)
This work provides a point of comparison between two anthracene-based bulky backbone carboxylic acid ligands and their corresponding complexes. The results indicate that the auxiliary ligands play an important role in the formation of frameworks of complexes 1–3, which together with the anthracene-based ligands and different anions may play an important role in both the structures and the luminescent properties of the complexes.
From the above descriptions, we can clearly see that the two anthracene-based ligands 9,10-dioxo-9,10-dihydroanthracene-1,5-dicarboxylic acid (H2 L 1) and 2-phenylquinoline-4-carboxylic acid (HL 2) exhibit variable coordination modes. In complex 1, the backbone of H2 L 1 locates in the same plane, and the dihedral angles between the carboxyl groups and backbone of the ligand are 86.0° and 92.1°. Therefore, H2 L 1 adopts a bis-monodentate bridging mode in 1. In complexes 2 and 3, because of N\( \cdots \)H small distances [2.442(3)–2.627(1) Å], the N atoms of HL 2 cannot coordinate to the metals. Hence, L 2 acts as a monocarboxylate ligand. The dihedral angles between the carboxyl group and ligand backbone are small (23.3°, 38.8° and 36.5°). Hence, HL 2 adopts rich coordination modes, namely monodentate or bidentate bridging in complex 2 and tridentate bridging in complex 3.
Spectroscopic properties
The IR spectra of complexes 1–3 show features attributable to each of their components [34]. A broad band at ca. 3,473–3,374 cm−1 is assigned to the O–H stretching of water [22, 34, 35]. The characteristic bands of the carboxylate groups appear in the usual region at 1,573–1,517 cm−1 for the antisymmetric stretching vibrations [ν as(COO−)] and at 1,463–1,407 cm−1 for the symmetric stretching vibrations [ν s(COO−)]. The Δν values [Δν = ν as(COO−) − ν s(COO−)] are 160 and 110 cm−1 for 1, 146 and 90 cm−1 for 2, 138 and 107 cm−1 for 3, in good agreement with their solid-state structures from the crystallographic analyses [22, 34, 35].
The solid-state luminescence spectra of complexes 1–3 as well as free ligands H2 L 1 and HL 2 were investigated at room temperature (see Fig. 5). While H2 L 1 and phen display weak luminescence in the solid state at λ max = 432 and 361/379 nm, respectively, upon excitation at λ ex = 380 and 310 nm, under the same experimental conditions, 1 exhibits intense purple emission at λ max = 432 nm upon excitation at 380 nm. Upon excitation with λ ex = 380 and 370 nm, complexes 2 and 3 exhibit intense emissions at 390 and 392 nm, respectively, which are close to the emission wavelengths of free HL 2, phen and pp (at 388, 361/379 and 410 nm, respectively) [15]. The similarity in the emission wavelengths between complexes 1–3 and the corresponding free ligands suggests that the emission of the complexes can be attributed to intraligand transfer π*–π transitions, namely ligand-to-ligand charge transfer (LLCT), similar to the reported results for other coordination polymers with anthracene-based backbones [36–38]. In addition, the enhanced luminescence intensities of complexes 1–3 compared to the free ligands can be explained by restricted vibration and rotation of the ligands within the complexes, which will decrease the contribution of radiationless deactivation [36–38].

Solid emission spectra of H2 L 1, HL 2 and 1–3 at room temperature (λ ex = 380 nm for H2 L 1, 279 nm for HL 2, 380 nm for 1, 380 nm for 2 and 370 nm for 3, respectively)
Magnetic properties and XRD results
The variable temperature magnetism of complex 2 was tested in the temperature range of 2–300 K at 2,000 Oe. The results are shown in Fig. 6 in the form of a plot of χ m T versus T (χ m is the molar magnetic susceptibility for two MnII ions). The value of χ m T at 300 K is 8.13 cm3 K mol−1, close to the χ m T value of two isolated MnII ions with g = 2, S = 5/2. The value of χ m T decreases along with temperature to 0.71 cm3 K mol−1 at 2 K. This phenomenon shows that an antiferromagnetic coupling interaction exists between the MnII ions in complex 2, in accordance with observations for other compounds with carboxyl bridged dinuclear MnII ions [39, 40]. The Curie–Weiss fitting of the data in the temperature range of 10–300 K for 2 gave values of C = 8.44 cm3 K mol−1 and θ = −6.38 K. The negative value of θ further confirms the existence of antiferromagnetic interactions in 2 [39, 40].

The plot of χ M T (filled square) and 1/χ M (filled circle) versus temperature for 2
To confirm whether the crystal structures are truly representative of the bulk materials, XRPD experiments have been carried out for complexes 1–3. The experimental and computer-simulated XRPD patterns are shown in Fig. 7. These patterns indicate that the bulk-synthesized materials and as-grown crystals can be considered homogeneous for all three complexes.

X-ray powder diffraction (XRPD) patterns of a for 1, b for 2 and c for 3
Conclusion
A series of metal–organic coordination compounds of manganese(II) and cadmium(II), generated from two anthracene-based bulky backbone ligands, together with phen or pp as auxiliary ligands, have been prepared. These complexes display diverse structures from binuclear to 1D zigzag chains. This work shows that the reactions of metals with the corresponding ligands are tunable through control of the reaction conditions. The auxiliary chelating ligands phen and pp play an important role in the formation of the frameworks of these complexes, preventing the formation of higher-dimensional structures. Intense luminescent emissions of the complexes were attributed to the LLCT of the corresponding ligands. Antiferromagnetic coupling interactions exist between the binuclear MnII ions in complex 2.
References
Sumida K, Rogow DL, Mason JA, McDonald TM, Bloch ED, Herm ZR, Bae TH, Long JR (2012) Chem Rev 112:724. doi:10.1021/cr2003272
Tian D, Chen Q, Li Y, Zhang YH, Chang Z, Bu XH (2014) Angew Chem Int Ed 53:837. doi:10.1002/anie.201307681
Lin ZJ, Lv J, Hong M, Cao R (2014) Chem Soc Rev. doi:10.1039/C3CS60483G
Schneemann A, Bon V, Schwedler I, Senkovska I, Kaskel S, Fischer RA (2014) Chem Soc Rev. doi:10.1039/C4CS00101J
Yan Y, Yang S, Blake AJ, Schröder M (2014) Acc Chem Res 47:296. doi:10.1021/ar400049h
Phan A, Doonan CJ, Uribe-Romo FJ, Knobler CB, O’Keeffe M, Yaghi OM (2010) Acc Chem Res 43:58. doi:10.1021/ar900116g
Liu J, Zhang HB, Tan YX, Wang F, Kang Y, Zhang J (2014) Inorg Chem 53:1500. doi:10.1021/ic402467g
Horie M, Suzaki Y, Hashizume D, Abe T, Wu T, Sassa T, Hosokai T, Osakada K (2012) J Am Chem Soc 134:17932. doi:10.1021/ja304406c
Liu MM, Hou JJ, Qi ZK, Duan LN, Ji WJ, Han CY, Zhang XM (2014) Inorg Chem 53:4130. doi:10.1021/ic5001232
He YP, Tan YX, Zhang J (2013) Inorg Chem 52:12758. doi:10.1021/ic4020256
Fang ZL, Yu RM, Wu XY, Huang JS, Lu CZ (2011) Cryst Growth Des 11:2546. doi:10.1021/cg200300x
Hou G, Bi L, Li B, Wu L (2010) Inorg Chem 49:6474. doi:10.1021/ic1001495
Park JH, Lee WR, Kim Y, Lee HJ, Ryu DW, Phang WJ, Hong CS (2014) Cryst Growth Des 14:699. doi:10.1021/cg401583v
Bu XH, Tong ML, Chang HC, Kitagawa S, Batten SR (2004) Angew Chem Int Ed 43:192. doi:10.1002/anie.200352024
Wang JJ, Liu CS, Hu TL, Chang Z, Li CY, Yan LF, Chen PQ, Bu XH, Wu Q, Zhao LJ, Wang Z, Zhang XZ (2008) CrystEngComm 10:681. doi:10.1039/b710209g
Wang JJ, Hu TL, Bu XH (2011) CrystEngComm 13:5152. doi:10.1039/c1ce05287j
Liu Y, He R, Wang F, Lu C, Meng Q (2010) Inorg Chem Commun 13:1375. doi:10.1016/j.inoche.2010.06.046
Tong B, Qiang JY, Xu YQ, Mei Q, Duan T, Chen Q, Zhang QF (2011) Inorg Chem Commun 14:1937. doi:10.1016/j.inoche.2011.08.029
Vogt FG, Williams GR, Johnson MN, Copley RCB (2013) Cryst Growth Des 13:5353. doi:10.1021/cg401232g
Wang JJ, Chang Z, Zhang AS, Hu TL, Bu XH (2010) Inorg Chim Acta 363:1377. doi:10.1016/j.ica.2010.01.043
Feng S, Xu R (2001) Acc Chem Res 34:239. doi:10.1021/ar0000105
Kendall JK, Shechter H (2001) J Org Chem 66:6643 and references therein. doi:10.1021/jo010404p
Bruker AXS (1998) SAINT software reference manual. Madison, Wisconsin
Sheldrick GM (1996) SADABS, Siemens area detector absorption corrected software. University of Göttingen, Germany
Sheldrick GM (1997) SHELXTL NT version 5.1. Program for solution and refinement of crystal structures. University of Göttingen, Germany
Wilkinson G, Gillard RD, McCleverty JA (eds) (1987) Comprehensive coordination chemistry, vol 5. Pergamon, Oxford
Zheng YQ, Lin JL, Chen BY (2003) J Mol Struct 646:151. doi:10.1016/S0022-2860(02)00615-4
Zhang Y, Li J, Zhu M, Wang Q, Wu X (1998) Chem Lett 1051. doi:10.1246/cl.1998.1051
Desiraju GR, Steiner T (1999) The hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Alekseyeva ES, Batsanov AS, Boyd LA, Fox MA, Hibbert TG, Howard JAK, MacBride JAH, Mackinnon A, Wade K (2003) Dalton Trans 475. doi:10.1039/B209931D
Li WW, Bing Y, Zha MQ, Li TH, Li X (2011) Acta Crystallogr Sect E 67:m1464. doi:10.1107/S1600536811039341
Janiak C (2000) J Chem Soc Dalton Trans 3885 and references therein. doi:10.1039/B003010O
Khlobystov AN, Blake AJ, Champness NR, Lemenovskki DA, Majouga AG, Zyk NV, Schröder M (2001) Coord Chem Rev 222:155. doi:10.1016/S0010-8545(01)00370-8
Nakamoto K (1986) Infrared and Raman spectra of inorganic and donor hydrogen bond coordination compounds. Wiley, New York
Deacon GB, Phillips RJ (1980) Coord Chem Rev 33:227. doi:10.1016/S0010-8545(00)80455-5
Balasubramanian K (1997) Relativistic effects in chemistry part A: theory and techniques; part B: applications. Wiley, New York
Valeur B (2002) Molecular fluorescence: principles and applications. Wiley-VCH, Weinheim
Miyasaka H (2013) Acc Chem Res 46:248. doi:10.1021/ar300102t
Li X, Cai Y, Fang Z, Wu L, Wei B, Lin S (2011) Cryst Growth Des 11:4517. doi:10.1021/cg200730k
Chang XH, LF LF, Hui G, Wang LY (2012) Cryst Growth Des 12:3638. doi:10.1021/cg300461v
Acknowledgments
The work was supported by the Joint Fund for Fostering Talents of National Natural Science Foundation of China and Henan province (No. U1204213), the National Natural Science Foundation of China (Nos. 21403006, 21304001 and 21301009), the Natural Science Foundation of Henan Province (No. 112102210371) and the Science and Technology Research Projects of Education Department of Henan province (12B150003).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Wang, JJ., Zhang, DJ., Zhang, RC. et al. Syntheses, crystal structures and properties of complexes with two anthracene-based bulky backbone ligands. Transition Met Chem 40, 69–77 (2015). https://doi.org/10.1007/s11243-014-9891-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-014-9891-0