
Abstract
Manganese(III) sulfato complexes cause the oxidative degradation of methylene blue and its partially and fully N-demethylated derivatives, azure B and thionine dyes, respectively, in sulfuric acid media. The reaction proceeds through a colored reactive organic radical generated in the first stage via one-electron oxidation of the starting material, leading to a mixture of N-demethylated and/or deaminated species. The rates of formation of the methylene blue and azure B radicals are much higher than those of their further decomposition, whereas the generation of the thionine radical is much slower than its immeasurably fast decay. The kinetics of decomposition of all three dyes and the methylene blue and azure B radicals were studied spectrophotometrically under isolation conditions at 298 K. The first stage of each reaction proceeds according to a second-order rate expression, being first order in the dyes and in the manganese(III) concentrations. Dependence of the pseudo-first-order rate constants on the oxidant concentration for the second stage exhibits a saturation effect under the applied conditions. It is postulated that electron transfer takes place between the [Mn(SO4)3]3− complex and the protonated form of the dye. The reactivity order of the dyes as determined from the second-order rate constants for the first reaction stage corresponds to the order of their HOMO energies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rich redox chemistry of manganese in aqueous solutions makes it essential for a number of biological systems involved in electron transfer reactions and for a variety of chemical processes. Thus, manganese(III) is employed in water splitting during the light reactions of green plant photosynthesis [1] and in enzymatic and nonenzymatic redox catalysis in mammalian systems [2–4]. It is also a powerful one-electron oxidant and a versatile, selective redox catalyst frequently used in laboratory and industrial organic synthesis involving oxygen transfer reactions to alkanes, alkenes and compounds containing nitrogen and sulfur atoms as well as carbon–carbon bond generation [5–16].
Previously, we have reported kinetic and mechanistic studies on oxidative degradation of some highly stable phenothiazine dyes of wide application, namely methylene blue (MB), azure B (AB) and thionine (TH), by another powerful one-electron oxidant, namely cerium(IV) in sulfuric acid media [17, 18]. The importance of manganese(III) in electron transfer processes has inspired us to use it as a degradation agent for these dyes. Sulfuric acid applied in these experiments is expected to affect the oxidation kinetics via complexation of the oxidant and protonation of the reductant. To the best of our knowledge, data on oxidation processes by manganese(III) in sulfuric acid media are so far limited to a study of the kinetics of l-glutamine oxidation by Made Gowda et al. [19].
Experimental
Methylene blue, azure B and thionine dyes, manganese(III) acetate dihydrate and manganese(II) sulfate monohydrate were purchased from Sigma-Aldrich and sulfuric acid from POCH. All the chemicals were of pro-analysis grade quality and used without purification. Solutions were made using water redistilled from alkaline permanganate. A stock solution of manganese(III) species was prepared by dissolving a weighed amount of its salt in a solution containing 0.1 M manganese(II) sulfate and sulfuric acid as required, at least 2 M.
Electronic spectra of manganese(III) species in sulfuric acid solutions presented in Fig. 1 were recorded with a Hewlett–Packard 8453 diode-array spectrophotometer equipped with constant temperature cell holder. Electronic spectra of reaction mixtures shown in Figs. 2, 3 and 4 were recorded using a home-made stopped-flow apparatus thermostatted with an external Julabo F25 cryostat and combined with the above-mentioned spectrophotometer. The “dead time” of this system was 0.1 s.

Electronic spectra of manganese(III) species in sulfuric acid solutions: [Mn(III)] = 0.01 M (a) and 2 × 10−4 M (b); [Mn(II)] = 0.1 M, T = 298 K, l = 1 cm

Spectroscopic changes during the methylene blue oxidative degradation by manganese(III). Conditions: [MB] = 2 × 10−5 M, [Mn(III)] = 2 × 10−4 M, [Mn(II)] = 0.05 M, [H2SO4] = 3.0 M, T = 298 K, l = 1 cm: a the radical formation, scans every 1 s, b the radical disappearance, scans every 0.8 s, cycle time increment by 10% after initial time of 6 s

Spectroscopic changes during the azure B oxidative degradation by manganese(III). Conditions: [AB] = 2 × 10−5 M, [Mn(III)] = 2 × 10−4 M, [Mn(II)] = 0.05 M, [H2SO4] = 3.0 M, T = 298 K, l = 1 cm: a the radical formation, scans every 1 s, b the radical disappearance, scans every 0.8 s, cycle time increment by 10% after initial time of 6 s

Spectroscopic changes during the thionine oxidative degradation by manganese(III). Conditions: [TH] = 2 × 10−5 M, [Mn(III)] = 2 × 10−4 M, [Mn(II)] = 0.05 M, [H2SO4] = 3.0 M, T = 298 K, l = 1 cm, scans every 1 s, cycle time increment by 10% after initial time of 10 s
TLC analysis of the reaction products was performed as before [18].
Kinetic measurements
Kinetics of the reactions were examined spectrophotometrically at T = 298 K using a home-built stopped-flow apparatus (thermostatted as mentioned above) that enables rate measurements down to t 1/2 = 0.01 s. All the kinetic runs were performed under pseudo-first-order conditions with the oxidant in at least tenfold excess. The concentration of the dyes was in the range of (1–6) × 10−5 M, usually 3 × 10−5 M. In all cases, the final concentration of Mn(II) was 0.05 M and the Mn(III) concentration was varied within the (0.3–3) × 10−3 M range. The concentration of H2SO4 was 3 M. Some preliminary kinetic experiments performed at H2SO4 concentrations varying within the 2.0–5.0 M range demonstrated the lack of substantial changes of the reaction rate with acid concentration. The rates of the two stages of the MB and AB degradation accompanied by strong absorbance changes were measured in separate experiments. The formation (first stage) and the disappearance (second stage) of the MB radical were followed at its λmax = 526 nm, Fig. 2. The destruction of the AB dye (first stage) and its radical (second stage) were monitored at 724 nm, being λmax of the dye and at 514 nm, being λmax of its semioxidized form, respectively, Fig. 3. In the case of the thionine--manganese(III) system, the second stage of the reaction was too fast to be measured; the first stage, i.e., the oxidation of the dye, was followed via the absorbance decrease within its characteristic absorption band, at λmax = 670 nm, Fig. 4. The timescales of the first steps of the MB, AB and TH oxidation were 0.2–2, 0.6–5 and 1.5–6 s, respectively; the timescales of decay of the MB and AB radicals’ were 120–125 and 24–45 s, respectively. Each kinetic run was repeated at least five times. Nonlinear least-squares analyses were performed on the absorbance versus time data obtained from kinetic runs for up to 95% of the overall absorbance changes. The relative standard errors of the single observed pseudo-first-order rate constants for the formation and the decomposition of the MB and AB radicals and for the TH disappearance were 0.5–2.0, 0.5–1.7 and 1–1.5%, respectively, and for the average values of the rate constants were 0.01–0.5%.
Results and discussion
Oxidation of phenothiazine dyes requires the use of a strong electron acceptor, E 0 > 1 V [20]. The standard redox potential for the \( {\text{Mn}}^{3 + }_{\text{aq}} /{\text{Mn}}^{2 + }_{\text{aq}} \) couple is reported as +1.51 V [21]. Complexation of manganese(III) in aqueous solutions significantly increases its stability and affects its redox properties. Diphosphate, citrate and edta, which are often used as manganese(III) stabilizing ligands, decrease the standard potential of the Mn(III)/Mn(II) couple to 1.15, 0.92 and 0.82 V, respectively [22], whereas sulfate barely changes the value [23]. Therefore, sulfato complexes of manganese(III) in sulfuric acid solutions are capable of oxidizing all of the studied dyes, presented in Scheme 1.

The oxidant, manganese(III), used in these studies, needs some comments. Hexaaquamanganese(III) examined in 1.0 M Mn(ClO4)2 and at variable HClO4 concentrations exhibits unusually high acidity (K a = 0.08 at 293 K), probably due to Jahn–Teller distortion (d 4 high-spin electron configuration) [24]. Mass action law considerations provide the commonly applied method for the preparation of manganese(III) solutions to avoid the hydrolysis and disproportionation of Mn(III) proceeding according to the simplified Eq. 1:
This equilibrium is shifted left in strongly acidic media in the presence of manganese(II). Thus, the stock solutions of manganese(III) always contain a large amount of manganese(II). Electronic spectra of the Mn(III)–Mn(II)–HClO4 system, carefully analyzed by Fackler et al. [25], show a characteristic band at 476 nm of apparent molar coefficient varying upon the perchloric acid concentration within the 54–115 M−1 cm−1 range due to the deprotonation of the [Mn(H2O)6]3+ cation and formation of its monohydroxo conjugate base. This band is attributed to the d–d transition in manganese(III) complexes distorted from O h symmetry (elongated along the z axis) due to the Jahn–Teller effect. The 5Eg ground and the higher-energy 5T2g terms are split into 5B1g and 5A1g, 5B2g and 5Eg terms, respectively. Thus, the single transition in O h symmetry is split into three; the observed band results from the unresolved 5B1g to 5B2g and 5Eg transitions [26]. More recently Jordan et al. [24] have also reported the presence of a higher energy transition with λmax at 300 nm and the molar absorption coefficient reaching its minimum at ca. 800 M−1 cm−1 for the lowest pH value. A remarkable optical property of the Mn(III)–Mn(II)–H2SO4 system investigated in this work is a continuous spectroscopic change with the sulfuric acid concentration, as shown in Fig. 1. The band at 483 nm (ε = 100 M−1 cm−1) in 2 M H2SO4 is shifted to 530 nm (ε = 160 M−1 cm−1) in 14 M H2SO4. The higher-energy, intense band at λmax 289 nm (ε = 3.2 × 103 M−1 cm−1), seen in Fig. 1b, is not well resolved until very high (14 M) H2SO4 concentration. To the best of our knowledge, information on electronic transitions in the UV range for the Mn(III)–Mn(II)–H2SO4 system has not been reported yet. The observed intensity increase and red shift of the lower energy band and formation of the higher energy, intense band, Fig. 1, cannot result from manganese(III) complexation by sulfates for at least two reasons; (i) the expected values of the stepwise stability constants (roughly estimated from the data for Ce(IV), Ce(III), Fe(III) and some M(II) sulfato complexes [27, 28] as K 1 > 100, K 2 and K 3 ca. 10 M−1) are high enough to guarantee at least 90% conversion of the manganese(III) into the [Mn(SO4)3]3− ion at 2 M H2SO4; (ii) the observed spectroscopic changes are too large to be caused by substitution of H2O ligands by SO4 2−. It seems reasonable to assume that the observed optical effect originates from the formation of mixed-valence manganese(III)–manganese(II) complexes bridged via sulfato ligands. The expected low value of the equilibrium constant for the reaction proceeding between two like-charged (anionic) species is consistent with a continuous increase in the mixed-valence complex content reflected in the observed continuous spectroscopic changes, Fig. 1. Although most of the manganese(II)–manganese(III) complexes belong to the Class I according to the Robin–Day classification [29], the found optical properties suggest rather a Class II case for the Mn(III)–SO4–Mn(II) mixed-valence complex. The changes within the visible spectrum range reported by Pinto et al. [23], namely an absorbance decrease with an increase in the sulfuric acid concentration within the 1–2 M range, can be explained by the transformation of the [Mn(H2O)5(OH)]2+ complex into its conjugate acid, [Mn(H2O)6]3+ ion.
Summing up, under the conditions applied in our kinetic experiments (3 M H2SO4), the mononuclear [Mn(SO4)3]3− anion is the predominant form of the oxidant used, though some amounts of the mononuclear [Mn(SO4)2]− ion and mixed-valence species of the Mn(III)–SO4–Mn(II) type are also present in solution.
Degradation of the phenothiazine dyes by manganese(III) could be examined only at sulfuric acid concentrations higher than 2 M because, as outlined above, at lower H2SO4 concentrations, the sulfato manganese(III) complexes are not stable. On the other hand, within the 2–5 M range, sulfuric acid concentration affects the reaction rate only slightly. The degradation process proceeds in two observed stages, as presented in Scheme 2.

The general reaction pattern of the phenothiazine dyes degradation
The organic radicals generated via one-electron transfer in the first stage are characterized by an intense absorption band within the visible spectrum at 526 and 514 nm for MB and AB, respectively, Figs. 2 and 3. The radical nature of these species, which were also generated using cerium(IV), was confirmed by their EPR spectra in our previous studies [17, 18]. The extremely high reactivity of the thionine radical makes its detection impossible. The second degradation stage, namely oxidation of the cationic radicals, leads to a mixture of thiazine dyes that can be separated by TLC; among these, we detected N-demethylated and/or deaminated derivatives of the starting dyes, mainly thionine and/or methylene violet (Bernthsen) and a few colored unidentified species, probably methyl thionoline, thionoline or thionol, as presented in Scheme 3 [30].

Structures of methylene violet (Bernthsen) (a), methyl thionoline (b), thionoline (c) and thionol (d) formed as the final reaction products
The kinetics of formation of the MB and AB radicals and their further oxidation were monitored in separate experiments whereas in the case of the TH system, only the first stage could be studied. Absorbance-time data collected under isolation conditions at λmax of the dyes or at λmax of their radicals fit the first-order rate expression very well and the values of the obtained pseudo-first-order rate constants summarized in Table 1 are practically independent of the chosen wavelength and the initial dye concentrations:
where S denotes the dye or its radical for the first- and the second reaction stages, respectively. Values of the pseudo-first-order rate constants of the first reaction stage plotted against the oxidant concentration give a linear profile, as shown in Fig. 5.

Dependence of the observed pseudo-first-order rate constants of the first reaction stage on manganese(III) concentration; [Mn(II)] = 0.05 M, [H2SO4] = 3.0 M, T = 298 K
The plots for formation of the MB and AB radicals pass through the origin within the error limits, which is consistent with the observed stability of these dyes in sulfuric acid and with the irreversibility of the overall process. In contrast, the plot obtained for the TH dye decomposition displays a small positive intercept that could result from the negligible reverse reaction, i.e., the oxidation of manganese(II) by the cationic thionine radical within the ionic pair. The calculated slope (Fig. 5) is a measure of the dye’s reactivity toward the trissulfatomanganate(III) complex at constant H2SO4 concentration, Eq. 3:
where HD2+ denotes the dye in its conjugate acid form, which predominates under the conditions applied in these kinetic measurements. The rate law derived from the reaction sequence outlined by Eq. 3 is of the form:
where at constant [Mn(III)]
If \( Q[{\text{Mn}}({\text{III}})] \ll 1 \), then \( k_{\text{obs}}^{\text{I}} = kQ[{\text{Mn}}({\text{III}})] \) and the obtained slope is the pseudo-second-order rate constant, being the product of the preequilibrium constant, Q, and the rate constant of the electron transfer within the ionic pair, k. The determined reactivity order (evaluated from the kQ products), HTH2+ (700 s−1 M−1) < HAB2+ (2,100 s−1 M−1) < HMB2+ (6,700 s−1 M−1), corresponds to the calculated HOMO energies of the protonated dyes; \( E_{{{\text{HTH}}^{2 + } }} \) = −14.62 eV, \( E_{{{\text{HAB}}^{2 + } }} \) = −13.90 eV, \( E_{{{\text{HMB}}^{2 + } }} \) = −13.71 eV, calculated at the HF/6-311++G(d,p) level [18].
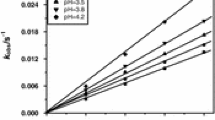
The second stage of the degradation process could be investigated only for the MB and AB dyes. As seen from Fig. 6, the dependence of the pseudo-first-order rate constant, \( k_{\text{obs}}^{\text{II}} \), on the manganese(III) concentration exhibits a saturation effect.

Dependence of the observed pseudo-first-order rate constants of the second reaction stage of methylene blue (a) and azure B (b) dyes degradation on manganese(III) concentration; [Mn(II)] = 0.05 M, [H2SO4] = 3.0 M, T = 298 K
Hence, in terms of the mechanism of this reaction stage analogous to that proposed for the first stage, Eq. 3, the value of the Q[Mn(III)] product, Eq. 5, is higher than 1. Then, from Eq. 5, the pseudo-first-order rate constant for the second stage \( k_{\text{obs}}^{\text{II}} = k \), where the constant k is interpreted as the first-order rate constant of the electron transfer within the ionic pair (cationic organic radical-anionic manganese(III) complex). Based on the collected data, Fig. 6 and Table 1, the values of k can be estimated as ca. 0.04 and 0.20 s−1 for the MB and AB systems, respectively, showing that the oxidation of AB radical is faster than that of MB. Lack of influence of the oxidant concentration on the \( k_{\text{obs}}^{\text{II}} \) value within the available manganese(III) concentration range could suggest disproportionation of the radicals. However, the decay of the radicals cannot result from this process, but rather from their oxidation by manganese(III), as evidenced by the following facts: (1) the disproportionation of the radical would regenerate the starting dye which is not observed; (2) the disproportionation process is characterized by second-order kinetics whereas a simple first-order reaction profile is well established; and (3) for the azure B—manganese(III) system, the rate dependence on [Mn(III)] is evident.
A remarkable feature of the present systems is the much lower value of the ionic pair formation constant for the starting dyes than those for their radicals. This can be attributed to their lower positive electric charge. In consequence, the second reaction stage is characterized by the first-order electron transfer rate constant, k, whereas the first reaction stage is described by the pseudo-first-order rate constant comprised of the electron transfer rate constant, k, and the preequilibrium constant, Q.
Conclusion
Distinct changes registered in the absorption spectra of sulfato manganese(III) complexes at [H2SO4] > 2 M, attributed to the formation of mixed-valence complexes, are not reflected in similarly large variations of the observed rate constants. This finding suggests that the mononuclear sulfato manganese(III) and the mixed-valence Mn(III)–SO4–Mn(II) type complexes have similar reactivities. Cerium(IV) and manganese(III), both of which are one-electron oxidants with similar standard redox potentials in sulfuric acid media, cause the oxidative degradation of the phenothiazine dyes by similar mechanisms and kinetics. The rate of reaction for cerium(IV) is only slightly higher [17, 18] than that for manganese(III) at the same concentrations of all other reactants. However, application of the latter one requires higher concentrations of sulfuric acid and additionally a large amount of manganese(II) to stabilize the manganese(III) species. Hence, manganese(III) is a much less convenient oxidant than cerium(IV) in these reactions.
References
Pecoraro VL, Hsieh WY (2008) Inorg Chem 47:1765–1778
Geiger R, Chattopadhyay S, Day VW, Jackson TA (2011) Dalton Trans 40:1707–1715
Riley DP (1999) Chem Rev 99:2573–2587
Rausaria S, Kamadulski A, Rath NP, Bryant L, Chen Z, Salvemini D, Neumann W (2011) J Am Chem Soc 133:4200–4203
Viciano-Chumillas M, Tanase S, Mutikainen I, Turpeinen U, de Jongh LJ, Reedijk J (2008) Inorg Chem 47:5919–5929
Giovannetti R, Alibabaei L, Puccarelli F (2010) Inorg Chim Acta 363:1561–1567
Kumar A, Goldberg I, Botoshansky M, Buchman Y, Gross Z (2010) J Am Chem Soc 132:15233–15234
Haber J, Matachowski L, Pamin K, Połtowicz J (2000) J Mol Catal A Chem 162:105–109
Kargar H (2011) Inorg Chem Commun 14:863–865
Brown MK, Blewett MM, Colombe JR, Corey EJ (2010) J Am Chem Soc 132:11165–11170
de Boer JW, Browne WR, Brinksma J, Alsters PL, Hage R, Feringa BL (2007) Inorg Chem 46:6353–6372
Liao S, List B (2010) Angew Chem Int Ed 49:628–631
Sheldon RA, Arends I, Hanefeld U (2007) Green chemistry and catalysis. Wiley-VCH Verlag, Weinheim
Bel-Daniel R, Weiner L, Neumann R (2002) J Am Chem Soc 124:8788–8789
Huang JW, Shi M (2005) J Org Chem 70:3859–3863
Wang YF, Toh KK, Ng EPJ, Chiba S (2011) J Am Chem Soc 133:6411–6421
Katafias A, Kita P, Wrzeszcz G (2007) Transition Met Chem 32:31–37
Katafias A, Fenska J (2011) Inter J Chem Kin 43:523–536
Rangappa KS, Chandraju S, Made Gowda NM (1998) Int J Chem Kin 30:7–19
Mills A, Wang J (1999) J Photochem Photobiol A Chem 123:123–134
Atkins P, Overton T, Rourke J, Weller M, Armstrong F (2010) Shriver and Atkins inorganic chemistry, 5th edn. Oxford University Press, Oxford
Klewicki JK, Morgan JJ (1998) Environ Sci Technol 32:2916–2922
Pinto I, Sherigara BS, Udupa HVK (1991) Analyst 116:285–289
Sisley MJ, Jordan RB (2006) Inorg Chem 45:10758–10763
Fackler JP Jr, Chawla ID (1964) Inorg Chem 3:1130–1134
Lever ABP (1984) Inorganic electronic spectroscopy, 2nd edn. Elsevier, Amsterdam
Sillen LG (1964) Stability constants of metal-ion complexes, Special Publication No. 17. The Chemical Society, London
Inczédy J (1976) Analytical applications of complex equilibria. Akadémiai Kiadó, Budapest
Gispert JR (2008) Coordination chemistry. Wiley-VCH, Weinheim
Marshall PN (1976) Histochem J 8:431–442
Acknowledgments
The authors would like to thank MSc Agnieszka Malinowska for participating in the experimental work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Katafias, A., Fenska, J. Oxidation of phenothiazine dyes by manganese(III) in sulfuric acid solution. Transition Met Chem 36, 801–809 (2011). https://doi.org/10.1007/s11243-011-9534-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-011-9534-7