
Abstract
The mer-[Ru(pic)3] isomer, where pic is 2-pyridinecarboxylic acid, undergoes base hydrolysis at pH > 12. The reaction was monitored spectrophotometrically within the UV–Vis spectral range. The product of the reaction, the [Ru(pic)2(OH)2]− ion, is formed via a consecutive two-stage process. The chelate ring opening is proceeded by the nucleophilic attack of OH− ion at the carbon atom of the carboxylic group and the deprotonation of the attached hydroxo group. In the second stage, the fast deprotonation of the coordinated OH− ligand leads to liberation of the monodentato bonded picolinate. The dependence of the observed pseudo-first-order rate constant on [OH−] is given by \( k_{{{\text{obs}}1}} = \frac{{k_{ + } k_{1} [{\text{OH}}^{ - } ] + k_{ + } k_{2} K_{1} [{\text{OH}}^{ - } ]^{2} }}{{k_{ - } + k_{1} + \left( {k_{ + } + k_{2} K_{1} } \right)[{\text{OH}}^{ - } ] + k{}_{ + }K_{1} [{\text{OH}}^{ - } ]^{2} }} \) and \( \left( {k_{{{\text{obs}}2}} = \frac{{k_{ca} + k_{cb} K_{2} [{\text{OH}}^{ - } ]}}{{1 + K_{2} [{\text{OH}}^{ - } ]}}} \right) \) for the first and the second stage, respectively, where k 1, k 2, k -, k ca , k cb are the first-order rate constants and k + is the second-order one, K 1 and K 2 are the protolytic equilibria constants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Transition metal picolinato complexes (where picolinate is pyridine 2-carboxylato anion, denoted as pic) are intensively studied because pic is used as a ligand for transportation of microelements to mammalian cells. The mer-[Cr(pic)3] isomer is widely applied as a synthetic source of biochromium [1–3], whereas its vanadium and ruthenium analogs are examined as potential diabetic and anticancer drugs, respectively [4]. At the turn of the twentieth century, several tris-, bis-, and monopicolinato complexes of ruthenium(II)/(III) have been synthesized and characterized [5–15]. They are attractive objects of study either as potential pharmaceuticals or as effective and selective stoichiometric or/and catalytic reagents toward oxidation of a variety of organic and inorganic substrates [9–11].
The mechanism of the chelate ligand liberation is still controversial, e.g., [16]. However, it has been found that if it is activated dissociatively, the chelate ring opening step is slower than the subsequent liberation of the monodentate bonded ligand. Hence, the intermediate complex is not accumulated in the system. In contrast, if the ring opening is faster than the ligand liberation, the intermediate accumulates and its spectral detection is possible. This is observed for the base hydrolysis of some chromium(III) pyridinocarboxylato complexes [17, 18]. Base hydrolysis of ruthenium(III) complexes has not received much attention mainly due to side reactions (e.g., oligomerization, disproportionation) resulting in complex kinetics. Some important features of base hydrolysis at Ru(III) centers are discussed in recent review articles (e.g., [19]) and can be summarized as follows: (1) the ligand substitution takes place invariably with the retention of configuration; (2) the reaction is activated dissociatively as judged by the large positive values of the apparent activation entropy, the positive values of the apparent activation volumes, and the linear dependence of the rate on pH; (3) stronger labilizing effect is caused by the amido than by the hydroxo group; (4) the rate of the ligand liberation is roughly two orders of magnitude lower for the aqua complexes than for their hydroxo derivatives. Similar magnitude of the labilizing effect is observed for chromium(III) aqua complexes converted into their hydroxo derivatives [20]. Molecular dynamics simulations based on empirically and quantum chemically calculated potentials [21] suggest an analogous square-pyramidal intermediate for ruthenium(III) and chromium(III) complexes because for both species, the HOMO orbital is occupied by a single electron.
In this paper, we describe results of our kinetic studies on the base hydrolysis of trispicolinatoruthenium(III), which was not examined earlier. The main aim of this work is the elucidation of the role played by protolytic reactions in the overall process.
Experimental
Unless otherwise stated, all chemicals were purchased from POCh (Gliwice, Poland) and as pro analysi grade, were not further purified; 2-pyridinecarboxylic acid 99% was purchased from Aldrich and RuCl3·nH2O from Merck. All experiments were performed using water redistilled from alkaline permanganate.
Synthesis of the mer-[Ru(pic)3]·H2O
mer-[Ru(pic)3]·H2O was prepared by the modified published procedure [11]. A solution of RuCl3·nH2O (2 mmol) in anhydrous ethanol (20 cm3) was refluxed for 3 h, filtered, and the volume of the dark green solution was reduced to 2–3 cm3. Then, an aqueous solution (20 cm3) of picolinic acid (20 mmol) was added and the mixture was stirred at 323 K till the color changed to orange (1 h). Yellow crystals were filtered off and recrystallized from hot water. Anal. Calculated for [Ru(pic)3]·H2O: C 44.0, N 8.4, H 2.9%; Found: C 45.0, N 8.0, H 2.9%.
Analyses
IR spectra were recorded on a Perkin Elmer Spectrum 200 instrument within the 4,000–400 cm−1 spectral range, using KBr pellets. The characteristic sharp and strong vibrations at 1,675, 1,282, 858, 763, 688, 468 cm−1 are consistent with the literature data [11]. The band at 1,675 cm−1 is assigned to the carbonyl stretching vibration of the coordinated carboxylato group in the picolinato ligand.
Single crystal structure determination was based on the diffraction data collected at ambient temperature using an Oxford Sapphire CCD diffractometer, Mo Kα radiation λ = 0.71073Å. The reflections have been measured with the ω-2θ method and the numerical absorption correction was applied [12]. The [Ru(pic)3]·H2O structure was solved by the direct methods and refined with full-matrix least squares procedure on F 2 (SHELX-97 [13, 14]).
The powder diffraction data collection was carried out using an X-ray Diffractometer Philips X’Pert PW 3040/60 with the copper radiation (K α = 1.5418Å) and the graphite monochromator on the diffracted beam. The measurements were provided in a range of 5–70°2θ with step of 0.02°2θ and exposition time 15 s. The theoretical diffraction pattern was simulated in PowderCell 2.3 (LIT). The mer-[Ru(pic)3]·H2O complex crystallizes in the monoclinic C2/c space group with all atoms found in general positions. The structure is identical to that reported by Barral et al. [14].
Thermal behavior of trispicolinatoruthenium(III) was investigated using a thermogravimetric method (TGA/DTA) over the temperature range 308–1,000 K at a ramp rate 100−1, applying SDT 2960 TA Instruments. The endothermic weight loss of 3.7% indicates one water molecule liberation (calculated weight loss 3.7%).
EPR spectra of the powdered mer-[Ru(pic)3]·H2O sample were recorded at room temperature with an X band (ca. 9.32 GHz) Radiopan EPR SE/X-2541 M spectrometer with a 100 kHz modulation. The microwave frequency was monitored with a frequency meter. The magnetic field was measured with an automatic NMR-type magnetometer. Although the spectrum was measured for a solid sample, three distinct signal are at g-values very close to those reported by Ghatak et al. [15] for liquid nitrogen frozen solution spectrum. UV–Vis spectra were measured with a Shimadzu UV-1601 PC spectrophotometer. Spectral data for the solution of the synthesized species in dimethylsulfoxide or acetonitrile are practically identical to those published earlier [15].
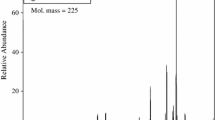
Electro-spray ionization mass spectrometric measurements were conducted with a Micromass Q-TOF instrument for the substrate aqueous solutions of pH 3 and 13. Results are shown in Fig. 1. The dominant peak at m/z = 491 and peaks of low intensity at m/z = 828, 958, 1,425 (Fig. 1a) are attributed to the monomeric trispicolinato complex and small amounts of dimer and higher aggregates present in the solution before the initiation of the reaction. Base hydrolysis leads to the formation of the [Ru(pic)2(OH)2]− ion identified by the peak at m/z = 426 and traces of some other species, Fig. 1b.

Electro-spray ionization mass spectra of [Ru(pic)3] a at pH = 3 and b at pH = 13; [RuIII] = 5 × 10−5 M
Aqueous solution of the ruthenium(III) complex
The mer-[Ru(pic)3]·H2O is scarcely soluble in water. It gives a pale yellow solution, from which, if concentrated up, yellow solid, characterized as the starting mer isomer, precipitates.
Kinetic measurements
The pale yellow solution becomes intense yellow due to formation of a strong band at λ = 448 nm. The reaction kinetics were studied spectrophotometrically (Fig. 2) using a HP 8453 diode-array instrument equipped with a HP 89090 Peltier temperature controller. The diode-array-type instrument is not recommended for kinetic measurements of ruthenium complexes because of their photosensibility. Nevertheless, data obtained using a Shimadzu UV-1601 PC spectrophotometer combined with an external Julabo F25 cryostat were practically the same.

Spectroscopic changes in UV–Vis region during the base hydrolysis of the mer-[Ru(pic)3]; [Ru(III)] = 5 × 10−5 M, [OH−] = 0.1 M, I = 1.0 M (Na+, ClO4 −, OH−), T = 298 K, scans taken every 3.2 s, cycle time increment by 10% after initial time of 50 s
The reaction progress was monitored as the absorbance changes at 236, 306, and 460 nm. The concentration of OH− was at least in 200-fold excess of that of the starting ruthenium(III). The ionic strength was kept at 1.0 M with NaClO4. The rate of the reaction was measured at five temperatures within the range 288–328 K. The kinetic data were analyzed with a double exponential function employing the Scientist program. Values of the pseudo-first-order rate constants (k obs1 and k obs2) are independent of the initial complex concentration. The relative standard errors for a single run were ca. 0.5%. Presented as an average of three kinetic runs k obs were reproducible within ±3%. Activation parameters were obtained from plots of ln(k/T) versus 1/T according to the Eyring equation.
Results and discussion
The solid trispicolinatoruthenium(III) complex was characterized by X-ray analysis, IR, and EPR spectroscopies. Some important structural data limited to the localization of the donor atoms are presented in Scheme 1 [14].

All the obtained data confirm the chelate mode of coordination of three picolinato ligands via the pyridine nitrogen and the carboxylato oxygen atoms, mer arrangement of the donor atoms, and presence of a single unpaired electron consistent with the low spin d 5 electron configuration of the central ion.
ESI MS results demonstrate that the reactant exists in aqueous solution mainly as the monomeric trispicolinato complex which is exceptionally inert within the pH range 0–11. In contrast, it easily undergoes one ligand substitution giving the [Ru(pic)2(OH)2]− anion in stronger alkaline media.
The overall spectral changes that occur upon mixing of the trispicolinatoruthenium(III) complex with OH− ions are shown in Fig. 2. The characteristic intense band present at 470 nm is attributed to LMCT transition, from OH− group to ruthenium(III). The acidification of the reaction mixture resulting in transformation of hydroxo to aqua ligand causes the disappearance of this band, Fig. 3.

Electronic spectrum of the base hydrolysis product; a in 0.1 M NaOH, b 0.1 M HClO4
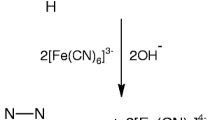
Since the reaction monitored as the absorbance increase at λmax of this band exhibits biphasic first-order kinetics, one can conclude that OH− ligand enters the inner coordination sphere in both stages of the reaction. The first stage is the chelate ring opening and the second one the-one-end bonded ligand liberation. Spectral changes in the UV region provide further information on the mechanism of the first reaction stage. The band at 248 nm ascribed to the electron transition in the coordinated picolinato ligand shifts toward lower energy and its intensity decreases. These changes are substantially larger for the second stage than for the first one. Deprotonation of the pyridine nitrogen atom of the free ligand is accompanied by a much larger band intensity decrease than the deprotonation of the carboxylic group [22]. Thus, it can be concluded that spectral changes in the UV region accompanying the base hydrolysis are consistent rather with the mechanism in which the reaction is initiated by the Ru–O bond breaking followed by the subsequent N-monodentato bonded picolinato ligand liberation, Eq. 1.
It is worth noting that hydrolysis of the examined Cr(III)-picolinato complexes proceeds via the Cr–N bond cleavage [22]. These findings are in accordance with the known preference of the chromium(III) center for O-donor atoms and ruthenium(III) for N-donor ones.
The pseudo-first-order rate constants for the chelate ring opening and the one-end bonded picolinato ligand liberation are summarized in Table 1.
A remarkable feature of the base hydrolysis of the mer-[Ru(pic)3] species is a strong rate acceleration with the [OH−] increase for the first reaction stage while for the second one, the reaction order with respect to [OH−] changes from one to zero for the lower and higher applied concentration limits, respectively. The dependences of the k obs1 and k obs2 on [OH−] are presented in Fig. 4.

Dependence of the k obs on [OH−] for two stages of the base hydrolysis of the mer-[Ru(pic)3]; a first stage; b second stage; [Ru(III)] = 5 × 10−5 M, I = 1.0 M (Na+, ClO4 −, OH−), T = 298 K
As seen, the data for the first stage fit a parabolic function:
where a is equal to zero within the error limits; for the second one the data fit a hyperbolic dependence:
The experimental data k obs1 and k obs2 versus [OH−] were fitted to Eqs. 2 and 3, respectively, applying a nonlinear least squares method.
The results for the first stage can be accounted for in terms of the reaction mechanism outlined in Scheme 2. The key concept is the nucleophilic attack of OH− ion on the carbon atom of the carboxylic group before the chelate ring opening process. The subsequent deprotonation of the attached hydroxo group results in the existence of the substrate in three forms (A, B, C) of reactivity increasing from A to C. Nucleophilic attack of the hydroxide ion on the coordinated ligands is well documented in the literature and now is “a common feature of the biochemical mechanisms” [16]. Results obtained by Yoshihara et al. [23] for the base hydrolysis of trisacetyloacetonatotechnetium(III) complex prove also that the attachment of the hydroxide ion to the carbon atom of the carbonyl group initiates the chelate ring opening process.

The rate law for the first stage derived from the proposed reaction mechanism is too complicated to be verified by the experimental kinetic data. The discussion of the obtained rate expression is possible only for three limiting cases. If the nucleophilic attack of the OH− ion represented by the rate constant k + is the rate determining step, then k obs1 is a linear function of OH− concentration, Eq. 4, what is not observed:
A faster than linear dependence of the pseudo-first-order rate constant, k obs1, on [OH−] found in this work is consistent with the assumption of fast preequilibria among A, B, C species before the rate determining steps represented by the rate constants k 0, k 1 and k 2 Eq. 5
or with the assumption of the steady state approximation for the reactive B and C intermediates, Eq. 6:
Equation 5 is simplified to a parabolic function if preequilibria \( \user2{A} \rightleftharpoons \user2{B} \rightleftharpoons \user2{C} \) are shifted toward A, i.e., A is the predominant form of the substrate. Then,
Similarly, if \( k_{ - } + k_{1} \gg k_{2} K_{1} [{\text{OH}}^{ - } ] \), Eq. 6 is simplified to
The parabolic dependences Eqs. 7 and 8 give kinetic evidence for the deprotonation of the hydroxo group attached to the carbon atom of the carboxylic group (Scheme 2). The cases described by these equations are experimentally indistinguishable from each other. Hence, the empirical kinetic parameters Eq. 2 are interpreted as: a = k 0 ≈ 0, b = Qk 1 and c = QK 1 k 2 Eq. 7 or as a = k 0 ≈ 0, b = k + k 1/k − + k 1 and c = k + k 2 K 1/k − + k 1 Eq. 8. In both cases the individual rate and preequilibria constants cannot be separated, so one can operate only with the apparent second- and third-order rate constants b and c, respectively.
It is seen that the OH− independent reaction path (A) is too slow to contribute to the base hydrolysis (k 0 ≈ 0). At 0.01 M [OH−], the lower limit of the applied concentration, about 90% of the product is formed from the B intermediate whereas at 0.5 M OH−, the highest employed [OH−], the path via C species contributes in about 80%.
Contrary to the first stage, namely the chelate ring opening process characterized by the profound rate acceleration caused by OH− ion, the rate of the second stage, the monodentato bonded picolinato ligand liberation, is only slightly affected by [OH−]. The data presented in Fig. 4 obey the hyperbolic function Eq. 3 satisfactorily, equivalent to the rate law Eq. 9 derived from the reaction model shown in Scheme 2:
where a ′ = k ca ≈ 0, \( b^{'} = k_{cb} K_{ 2} \) and c ′ = K 2. The mechanism for the second stage of the base hydrolysis assumes the fast deprotonation of the coordinated OH− ligand followed by the rate limiting liberation of the monodentato bonded picolinate. It can be verified by comparison of the value of the kinetic parameter \( K_{2} = K_{\text{a}} /I_{\text{w}} \) where I w is the ionization product of water and K a is the acidity constant for the monohydroxoruthenium(III) complex. As calculated from the data shown in Fig. 4 , the pKa 13.1 is in a good agreement with the values of 13–14 given in the literature for the deprotonation of the coordinated hydroxo ligand in some ruthenium(III) complexes [24].
The temperature dependence of the k obs2 value was found for the highest employed [OH−] = 0.5 M (Table 1). Under such conditions \( K_{2} [{\text{OH}}^{ - } ] \gg 1 \) and therefore, from Eq. 9, k obs2 = k cb . Thus, the calculated activation parameters, ∆H # = 60 ± 1 kJ mol−1 and ∆S # = −98 ± 4 J K−1 mol−1, describe liberation of the picolinato ligand from the C complex, Scheme 2.
Summing up, in spite of the theoretical resemblance of the activation mode for ruthenium(III) and chromium(III) ligand substitution, there are some important differences in kinetic behavior of their trispicolinato complexes: (1) acid catalyzed hydrolysis is observed only for the Cr(III) complex [22]; (2) the strong rate dependence on OH− concentration for the base hydrolysis of the mer-[Ru(pic)3] species contrasts with lack of such influence for its chromium(III) analog (E. Kita, unpublished data); (3) the rates of the first and second stage of the picolinato ligand substitution for the chromium(III) complex are similar; the value of the k obs1 is only 2–3 times higher than that of the k obs2; for the ruthenium(III) species, instead the chelate ring opening is faster than the further one-end bonded ligand liberation by more than one order of magnitude. One more essential difference between the systems under consideration is formation of the intermediate products with the monodentato N- and the monodentato O-bonded picolinate as a consequence of the OH− ion attack on the carboxylato group coordinated to the ruthenium(III) and directly on the chromium(III) center, respectively. A similar unexpected difference is observed also for the base hydrolysis of trisacetyloacetonatoruthenium(III) and technetium(III) complexes [23].
References
Korea SK, Jpn Kokai Tokkyo Koho JP 08, 291, 190 [96, 291, 190] (ClC07F11/00), 5 Nov 1996, KR Appl 9,508,651, 13 Apr 1995
Kingry KF, Royer AC, Vincent JB (1998) J Inorg Biochem 72:79
Evans GW, Johnson EC (1981) J Nutr 111:68
Jones CJ, Thornback JR (2007) Medicinal applications of coordination chemistry. RSC Publishing, Cambridge
Ellis RM, Quilligan JD, Williams NH, Yandell JK (1989) Aust J Chem 42:1
Ghatak N, Chakravarty J, Bhattacharya S (1995) Transit Met Chem 20:138
Sengupta P, Dinda R, Ghosh S, Sheldrick WS (2001) Polyhedron 20:3349
Ghatak N, Chakravarty J, Bhattacharya S (1996) J Chem Sci 108:286
Chatterjee D, Mitra A (2000) Inorg Chem Commun 3:640
Chatterjee D, Sengupta A, Mitra A, Basak S (2006) Inorg Chem Commun 9:1219
Chatterjee D, Sengupta A, Mitra A (2007) Polyhedron 26:178
Rachford AA, Petersen JL, Rack JJ (2007) Dalton Trans 3245
Mitra KN, Choudhury S, Goswami S, Peng S-M (1997) Polyhedron 16:1605
Barral MC, Jiménez-Aparicio R, Royer EC, Saucedo MJ, Urbanos FA, Gutiérrez-Puebla E, Ruíz-Valero C (1991) J Chem Soc Dalton Trans 1609
Ghatak N, Chakravarty J, Bhattacharya S (1995) Polyhedron 14:3591
Jordan RB (2007) Reaction mechanisms of inorganic and organometallic systems, 3rd edn. Oxford University Press, New York
Kita E, Marai H (2009) Transit Met Chem 34:585
Kita E, Marai H, Lisiak M, Jasiński M, Drewa T (2010) Transit Met Chem 35:177
Richens DT (2005) Chem Rev 105:1961
Helm L, Merbach AE (2005) Ibid 105:1923
Rotzinger FP (2005) Ibid 105:2003
Kita E, Łączna M (2001) Transit Met Chem 26:510
Mutalib A, Omori T, Yoshihara K (1993) J Radioanal Nucl Chem 170:67
Liu F, Concepcion JJ, Jurss JW, Cardolaccia T, Templeton JT, Meyer TJ (2008) Inorg Chem 47:1727
Acknowledgments
This work was financially supported by Authorities of N. Copernicus University through grant No. 356-Ch. The authors wish to express their gratitude to the Amicus Universitatis Nicolai Copernici Foundation for the financial support, Dr. T. Muzioł for X-ray studies, and MSc. B. Biri for MS measurements.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Impert, O., Katafias, A., Kita, P. et al. Base hydrolysis of mer-trispicolinatoruthenium(III): kinetics and mechanism. Transition Met Chem 36, 761–766 (2011). https://doi.org/10.1007/s11243-011-9530-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-011-9530-y