
Abstract
No data is available about pharmacological secondary prevention of superficial vein thrombosis (SVT) despite 10–15% of patients develop venous thromboembolic complications at 3–6 months after an adequate treatment of the acute phase. To verify efficacy and safety of mesoglycan in secondary prevention of SVT recurrence and venous thromboembolic complications. Phase III multicenter, double-blind, randomized, superiority trial comparing mesoglycan 50 mg bid vs placebo in consecutive patients with a SVT extended at least 5 cm, after the initial 45-day treatment course with fondaparinux 2.5 mg once-daily. Primary efficacy outcome: SVT recurrence/extension, symptomatic venous thromboembolism (VTE), asymptomatic proximal deep-vein thrombosis, death. Primary safety outcome: major bleeding. We hypothesized a 12-month 15% incidence of the primary efficacy outcome in placebo group and a 50% risk reduction in mesoglycan group. A bilateral log-rank test with a sample of 650 patients (randomization 1:1) reach a 90% power, with an α-error of 0.025, of detecting a 7.0% difference (HR = 0.51) after 12 months of treatment, considering a 10% patients drop-out. At deadline (December 31, 2022) 570 patients have been randomized (10% drop rate). Mean age was 63.9 years, 58.8% were women. SVT involved great saphenous vein in 69.3%, small saphenous vein in 13.1%, and collaterals in 17.6% of patients. SVT was the first event in 61.7%, a recurrence in 38.3%, provoked in 50.2% and unprovoked in 49.8%. Patients not experiencing a primary outcome, or not retiring their consent will be followed up to December 31, 2024 when the final data analysis will be performed
ClinicalTrials.gov: NCT03428711.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Highlights
-
Superficial vein thrombosis is frequently associated to venous thromboembolism
-
No data is available about secondary prevention of superficial vein thrombosis
-
Mesoglycan could prevent recurrences with a long-term treatment versus placebo
-
Lacking data on the natural history of the superficial vein thrombosis will be recorded
Introduction
Superficial vein thrombosis (SVT) is a relatively common clinical condition characterized by a combination of thrombosis, inflammation, and pain along the course of a superficial vein [1]. A recent epidemiological community study clearly showed an incidence of SVT of 0.64% per year, which is six times higher than the incidence of venous thromboembolism (VTE), with an annual diagnosis rate ranging from 0.04 [95% CI, 0.00–0.10%] in men aged 18–39 years to 2.2% [95% CI,1.59–2.78%] in women aged > 75 years [2].
Considered a benign disease for a long time, SVT was recently shown to be associated with deep vein thrombosis (DVT) and symptomatic pulmonary embolism (PE) in up to 25% and 13% of cases, respectively [2, 9]. Proximal SVT (thigh) was also found to be associated with asymptomatic PE in up to 33% of cases [2, 9].
Patients with SVT and obesity, cancer, male sex, personal or family history of VTE, or severe chronic venous insufficiency are at increased risk of developing short- and long-term complications of VTE [10,11,12,13,14].
Indeed, recognizing a strong association between SVT and VTE in the last 10–15 years significantly modified the perceived risk of the former and its therapeutic approach, especially in the acute phase. In this respect, only fondaparinux (2.5 mg OD for 45 days) was shown to significantly reduce VTE complications, SVT extension, or recurrence by approximately 80% compared to placebo; while studies with low molecular weight heparins gave conflicting or less conclusive results [9, 10, 15, 16]. Consequently, fondaparinux (2.5 mg OD for 45 days) currently represents the first choice for the acute phase treatment of patients with SVT of the lower extremities [17].
Once completed the treatment of the acute phase, several studies have shown the utility of a treatment extension, either with vitamin K antagonists, or with direct oral anticoagulants, or low-molecular-weight heparins, or even aspirin or sulodexide as compared to no treatment in preventing VTE recurrence in patients with a first DVT and/or PE event [18,19,20,21,22].
On the contrary, no data is available on the need for extended treatment / secondary prevention of thromboembolism after SVT. However, despite adequate anticoagulant treatment in the acute phase, the incidence of VTE complications is as high as 15% at 3–6 months from the index event [3, 5, 15].
Mesoglycan is a complex mucopolysaccharide formed by a blend of natural glycosaminoglycans, mainly heparin-sulfate and dermatan-sulfate. Pharmacokinetic animal studies (in rats and monkeys) with tritium-labeled mesoglycan showed peak plasma levels of mesoglycan between 30 and 120 min after oral administration [23, 24]. The steady-state condition was maintained for up to 7 h; presumably due to a slow-release of the drug, after absorption by the gut-wall [23, 24]. To the contrary, peak plasma levels were recorded almost instantaneously after intravenous administration, with a subsequent rapid concentration fall within 60 min, following a biphasic course [23, 24].
The pharmacological activity of mesoglycan is expressed at the endothelial and subendothelial level, and is mainly due to the presence of heparan and dermatan sulfate, which are physiological constituents of the vessel wall.
Concerning its pharmacological effects, mesoglycan is a mild thrombin inhibitor with complementary action mediated by antithrombin and heparin-cofactor II, and is also profibrinolytic, stimulating the tissue plasminogen activator. It also exerts an antiatherogenic effect, via the inhibition of platelet adhesion, the stimulation of enzyme lipoprotein lipase, and the inhibition of the proliferation of smooth muscle cells of the tunica media; and an antiinflammatory effect via reduction of plasmatic concentration of several different cytokynes (such as MMP2, MMP9, IL6, TNFα, ICAM1, VCAM1) [25,26,27,28,29,30,31]. Furthermore, limited to the venous side of the circulatory system, mesoglycan is able to restore the physiological properties of the selective barrier represented by capillary endothelium, restoring flow-mediated vasodilation in subjects with endothelial dysfunction, repairing venous wall damage caused by chronic venous insufficiency, and accelerating the healing of venous ulcers [25,26,27,28]. Mesoglycan does not significantly influence coagulation parameters. In the literature there is only a small study showing a statistically significant decrease in fibrinogen plasma concentrations and an improvement in erythrocytes filterability following mesoglycan treatment, but no effect was found on prothrombin time, activated partial thromboplastin time or antithrombin levels [32].
While the efficacy of mesoglycan in preventing recurrence after a (first) episode of DVT is documented, there is no published evidence on the efficacy and safety of mesoglycan for the prevention of recurrence in patients with SVT of the lower limbs [33, 34].
Aim of the study
The aim of the METRO (Mesoglycan for secondary prevention of superficial vein thrombosis), a randomized controlled and double-blind study, is to evaluate the usefulness of mesoglycan for the secondary prevention of VTE, after the initial 45-day course of subcutaneous fondaparinux (2.5 mg OD), in patients with SVT of the lower limbs.
Materials and methods
Design of the study
The METRO study is a multicenter, phase 2, randomized, double-blind, superiority trial that compares mesoglycan 50 mg BID with a matching placebo. Using a placebo as a comparator is justified by the absence of scientific evidence on the efficacy and safety of any treatment in this setting. The METRO study is conducted in 19 Italian Units of Vascular Medicine, all with significant experience in the management of VTE. The Coordinating Centre will be the General Medicine Unit and the Thrombotic and Haemorrhagic Disorders Unit at the Department of Medicine—Padua University Hospital, Italy. The study protocol has been reviewed and approved by the Ethics Committee of the Padua University Hospital, Italy, and by those of each participating center.
Before inclusion, patients/participants are requested to sign a written informed consent form.
An independent data safety monitoring board (DSMB), blinded to the patient's treatment allocation, formed by three highly qualified clinicians in the field of VTE, will adjudicate all efficacy events, monitor patients' safety, and vouch for the data quality.
Study population and randomization
Subjects of both sexes, > 18 years of age, with objectively documented SVT of the lower limbs, initially extended for at least 5 cm, who had completed the standard 45-day course with fondaparinux 2.5 mg OD, were asked to participate in the study. Patients could only be included if they consented to participate within two weeks from treatment withdrawal. The complete list of inclusion and exclusion criteria is reported in Table 1.
After signing the informed consent, eligible subjects will complete the screening visit. Only patients who meet the inclusion and exclusion criteria will be included.
The included patient will first undergo bilateral color-coded Doppler ultrasound (CCDU) of the lower limbs to exclude concomitant involvement of the deep venous system or an extension of the index SVT. Furthermore, blood sampling for alanine-aminotransferase, aspartate-aminotransferase, creatinine, complete blood count, prothrombin time, activated partial thromboplastin time, and C-reactive protein will be performed.
A computer-based centralized randomization, with 1:1 allocation between mesoglycan and placebo, and random variable size blocks (4 to 6 units), stratified for age (< 60 y, > = 60 y), and gender (F/M), will be run through a dedicated application (REDCAp). Patient recruitment is expected to be completed in 24 months.
Treatment
Patients will receive a hard capsule of 50 mg mesoglycan or a matching placebo twice daily for 12 months. Treatment packages will be labeled with a bare numerical code without reference to the content. The match between the numerical code and the package content will only be known to the packaging manager. Patients, investigators, the steering committee, and DMSC will be blinded to treatment group allocation. The blinding seal will only be opened after the database lock to allow the analysis of the study results, except for in the case of a medical emergency, for which it is considered essential to know the type of treatment.
Each participating center will keep a printed drug accountability form for each patient included in the study (drug amount delivered, consumed or returned).
Patient flow
Medical control visits will be scheduled 3, 6, 9, and 12 months after randomization for every included patient, during which a complete evaluation of their clinical status, compliance with treatment, concomitant treatments and adverse events will be performed. At each visit, patients will also receive the revised Venous Clinical Severity Score (rVCSS), which accounts for changes in disease severity over time and in response to treatment, and the VEINES/Sym-QoL questionnaire, a disease-specific quality of life instrument for chronic venous disorders of the leg [34,35,36,37,38,39]. A CCDU examination and a blood sample will also be scheduled for the 12 month visit.
At the end of the 12-month treatment cycle, patients will enter another 12-month clinical follow-up period, during which they should not receive any experimental treatment; for a total study time of 24 months. A final visit will be scheduled 24 months after randomization in patients who complete the study course.
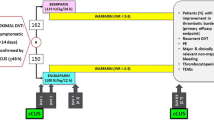
Patients who meet exclusion criteria at any time during the study, become pregnant, do not comply with study purposes or withdraw their consent will be censored. Patients who withdraw experimental treatment will continue to observe according to the study protocol until the end of the study. The planned flow of the patient is shown in Fig. 1.

Flow of the patients through the study
Study outcomes
The primary efficacy outcome of the study will be an objectively documented symptomatic recurrence or extension of SVT, symptomatic DVT (proximal or distal), asymptomatic proximal DVT, symptomatic pulmonary embolism (fatal / nonfatal), or death, whichever comes first, during the treatment period.
The secondary efficacy outcome will be: the occurrence of asymptomatic recurrence, extension, or asymptomatic distal DVT; the rVCSS score and the VEINES / Sym-QoL questionnaire; the impact of elastic compression therapy on SVT treatment; the association between CRP levels and primary efficacy outcomes; the natural history of SVT in patients allocated to placebo; the evaluation of the prolonged protective effect of mesoglycan after the first 12 months of treatment.
The safety outcome will be the incidence of major bleeding, as defined by the International Society on Thrombosis and Hemostasis (ISTH) criteria: fatal and/or symptomatic bleeding in a critical area or organ (e.g., intracranial, intraspinal, intraocular, retroperitoneal, intraarticular, pericardial, intramuscular with the associated compartmental syndrome); and/or in the case of clinically overt bleeding associated with a hemoglobin decrease of at least 20 g/L or requiring transfusion of 2 or more units of packed red blood cells [40].
Outcome assessment
Patients will receive detailed instructions on how to contact their referral center if symptoms suggestive of efficacy or safety events occur during the course of the study (treatment period and clinical follow-up).
Patients with suspected recurrent SVT or DVT will undergo CCDU. As previously described, whole-leg CCDU will be performed according to a standardized protocol [41]. Briefly, the proximal venous system of the lower limbs (common, superficial, and deep femoral veins, and popliteal vein to the trifurcation) is examined first with the patient lying supine; then the distal venous network (tibio-peroneal and muscular veins) is scanned, with the patient sitting. We do not routinely investigate the anterior tibial veins. Therefore, vein incompressibility is the only diagnostic criterion used to rule in proximal and/or distal DVT and SVT. Patients with symptoms suggestive of PE are investigated with Computed Tomography Pulmonary Angiography (CTPA) or Pulmonary Ventilation (V)and Perfusion (Q) scan. PE is adjudicated in the presence of an intraluminal filling defect involving the principal, lobar, or segmental branches of the pulmonary artery. If the patient dies, the cause of death is adjudicated as "likely" or "unlikely" due to PE based on autopsy findings, if available, or on clinical grounds.
Data Collection
Data will be recorded on an “Electronic Data Capture” (EDC) system, based on the “Research Electronic Data Capture” online platform (REDCap, produced and distributed by Vanderbilt University and “REDCap Consortium”), in compliance with article n. 13 of the GDPR (EU 2016/769) [42].
The personal data of the subjects enrolled will be treated with maximum confidentiality, according to the terms of Italian law (DL 211/2003 and subsequent amendments and additions) and GDPR [Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016].
In particular, the subject's personal data will be known only to the principal investigator and his collaborators who follow the patient and collect the treatment consent. Each enrolled subject will be distinguished only by a unique numeric identifier. The use and maintenance of the REDCap platform are guaranteed by an administrator who manages the user's privileges with a flexible and granular authorization system. REDCap applies the permissions granted to each user who connects via a web browser and the SSL encryption protocol. REDCap allows a complete "audit" of the procedures performed by each user by logging all operations on the data, including viewing and exporting. The operational control log stores the date, time, and operating behavior of the user, allowing a complete review and remote monitoring of the clinical study if necessary. All users accessing the EDC platform (the investigators, the members of their staff, and the staff of the Promoter) must complete a training event to increase the reliability, quality, and integrity of the data recorded in the EDC platform. REDCap implementations allow compliance with the most common industry standards and EMA requirements (GCP, Privacy-IT: DL 211/2003 and subsequent amendments and additions, GDPR, and FDA (21-CFR2-Part 11).
Statistical analysis and sample size
From the available data in the literature we hypothesize a risk of the occurrence of the primary efficacy outcome in the placebo group at the end of the treatment of 15% (that is, a cumulative proportion of 85% of subjects without events) and a 50% decrease in risk in the mesoglycan group (HR = 0.5) [3, 5, 9].
A two-sided log-rank test with an overall sample size of 650 subjects (of which 325 are in the placebo reference group and 325 in the mesoglycan treatment group) achieves a power of 90%, at a significance level (alpha) of 0.025, to identify a difference of 7.2%, between 15.0 and 7.8% (the proportions of subjects with primary endpoint in the placebo and mesoglycan group, respectively) after 12 months of treatment. This corresponds to a hazard ratio (HR) = 0.50. Subjects will be enrolled in the study for 24 months, and 50% of enrollments will be completed in month 12. The calculation considers the loss of subjects enrolled during follow-up of 10.0% in the placebo group and 10.0% in the mesoglycan group. 650 subjects will be enrolled and randomized 1:1 in the two placebo and mesoglycan arms.
This number seems large enough to guarantee the study an adequate “power” (> 0.80) even if the risk of observing the primary endpoint in the placebo group (hazard rate control) is the difference in efficacy between the two treatments (hazard ratio) that will deviate significantly from those hypothesized.
However, an “interim-analysis” is prepared when 3/4 of the subjects (n = 488) are enrolled so that the “Data and Safety Monitoring Committee” can reformulate the number of subjects to be enrolled (“resampling”) to reach the preset study power (90%) if the number of primary endpoint events observed up to then deviates significantly from the expected for the two groups, i.e., if the HR differs from the estimated one (50%) exceeded 5% in absolute value (a lower deviation seems tolerable based on the above simulation).
The alpha error is, therefore, conservatively set at 0.025 (O'Brien-Fleming method).
For the interim analysis, a tabular description of the frequency of the primary efficacy endpoint will be provided to the Committee, stratified by treatment group but without its label (i.e., without identifying the nature of the treatment assigned to each group).
Statistical considerations
All variables will be descriptively analyzed with the appropriate statistical methods: for categorical variables using frequency tables and for continuous variables with sample statistics (e.g., mean, median, standard deviation, minimum and maximum value, 25° and 75° quartile).
Unless otherwise specified, all statistical tests will be 2-tailed and at a 5% significance level.
All the primary and secondary efficacy and safety analyses will be performed in the modified intention-to-treat (ITT) population, including all randomized subjects who received at least one dose of the study drug.
Further supporting analysis will be performed in the per-protocol population, which will include all randomized subjects who were complying with treatment, that is, taking at least 65% of the intended dose units of the study drug.
Subjects who are randomized and who have taken any amount of study drug will not be replaced for any reason. Subjects who will be randomized but withdraw their consent before taking the study drug may be replaced, although they will not lose their unique identifier.
Subjects who leave the observation without manifesting the endpoint will be considered censored, participating in the risk during the observation period. For subjects who do not appear for clinical evaluations at 12 and 24 months of observation, every effort will be made to know a possible outcome of interest for the study, making them evaluable for endpoints.
Treatment compliance
Compliance with oral treatment can be used as a stratification variable (< 65% and > = 65%) and defined as compliance = the total number of tablets actually taken / the total number of tablets expected*100.
Final analysis of the primary endpoints
The primary cumulative efficacy endpoint will be the occurrence of the first event among the event types considered. The hypothesis of superiority of the primary cumulative efficacy endpoint will be tested by multivariate Cox proportional hazards regression using as covariates the assigned drug, age at randomization, sex, compliance with treatment, and the additional risk factors for DVT and DVT/PE found to be significantly associated in the univariate analysis (between obesity, varicose veins, cancer, standing or prolonged immobilization, recent surgery, severe infections, prior DVT/PE, thrombophilia, chronic heart failure NYHA III-IV, use of estrogen-progestogens). All variables participating in the model will be tested for the assumption of proportionality of the risks with the usual graphical method. In addition, a significant subset of variables will be chosen with the Wald method and in a “forward stepwise.”
The crude and adjusted cumulative proportion of event-free subjects over follow-up in the two treatment groups will be described with Kaplan–Meier plots.
Final analysis of secondary endpoints
For the analysis of the secondary efficacy endpoints, the following will be used:
-
1.
the Kaplan–Meier plot of the cumulative occurrence over time and the log-rank test to compare the two treatment arms,
-
2.
for the rVCSS scores, analysis of variance for repeated measures will be used (all available assessments for within-subject effects and the 2 treatment groups as between-subjects effect).
-
3.
in the case of the VEINES/Sym-Qol scores, the analysis of variance for repeated measures will be used (all evaluations available for the within-subject effect and the 2 treatment groups as the between-subjects effect).
-
4.
The impact of elastic compression therapy in the various endpoints will be analyzed by inserting it as a stratification factor in the related analyzes.
-
5.
The predictive contribution of CRP levels (subdivided by tertiles) to the primary efficacy endpoint will be analyzed by insertion as a stratification factor in relative analyzes.
Final analysis of the security endpoints
Analysis of the primary safety endpoint will be performed with the continuity adjusted chi-square test.
Standard summary of safety data related to adverse events and significant changes in laboratory data will also be provided.
Data availability
The data that support the findings of this study are not openly available due to reasons of sensitivity and are available from the corresponding author upon reasonable request.
References
Janson JN, Ascher E, De Pippo P, Lorensen E, Scheinman M, Yorkovich W et al (1998) Saphenous vein thrombophlebitis(SVT): a deceptively benign disease. J Vasc Surg 27:677–680
Frappe P, Buchmuller-Cordier A, Bertoletti L, Bonithon-Kopp C, Couzan S, Lafond P, Leizorovicz A, Merah A, Presles E, Preynat P, Tardy B, Decousus H, the STEPH Study Group (2014) Annual diagnosis rate of superficial-vein thrombosis of the lower limbs: the STEPH community-based study. J Thromb Haemost 12:831–838
Decousus H, Quere I, Presles E, Becker F, Barrellier MT, Chanut M, Gillet JL, Guenneguez H, Leandri C, Mismetti P, POST (Prospective Observational Superficial Thrombophlebitis) Study Group (2010) Superficial venous thrombosis and venous thromboembolism: a large prospective epidemiologic study. Ann Intern Med 152:218–224
Kitchens CS (2011) How I treat superficial vein thrombosis. Blood 117:39–44
Galanaud J-P, Genty C, Sevestre M-A, Brisot D, Lausecker M, Gillet JL, Righini M, Leftheriotis G, Bosson JL, Quere I, OPTIMEV SFMV Investigators (2011) Predictive factors for concurrent deep vein thrombosis and symptomatic venous thromboembolic recurrence in case of superficial venous thrombosis. OPTIMEV study Thromb Haemost 105:31–39
Decousus H, Frappe P, Accassat S, Bertoletti L, Buchmuller A, Seffert B, Merah A, Becker F, Quere I, Leizorovicz A (2012) Epidemiology, diagnosis, treatment and management of superficial-vein thrombosis of the legs. Best Pract Res Clin Haematol 25:275–284
Verlato F, Zucchetta P, Prandoni P et al (1999) An unexpectedly high rate of pulmonary embolism in patients with superficial thrombophlebitis of the thigh. J Vasc Surg 30:1113–1115
Di Minno MND, Ambrosino P, Ambrosini F, Tremoli E, Di Minno G, Dentali F (2016) Prevalence of deep vein thrombosis and pulmonary embolism in patients with superficial vein thrombosis: a systematic review and meta-analysis. J Thromb Haemost 14:964–972
Leizorovicz A, Becker F, Buchmuller A, Quere I, Prandoni P, Decousus H, CALISTO Study Group (2013) Clinical relevance of symptomatic superficial-vein thrombosis extension: lessons from the CALISTO study. Blood 122:1724–1729
Cosmi B, Filippini M, Campana F, Avruscio G, Ghirarduzzi A, Bucherini E, Camporese G, Imberti D, Legnani C, Palareti G, STEFLUX Investigators (2014) Risk factors for recurrent events in subjects with superficial vein thrombosis in the randomized clinical trial SteFlux (Superficial Thromboembolism Fluxum). Thromb Res 133:196–202
Scott G, Mahdi AJ, Alikhan R (2015) Superficial vein thrombosis: a current approach to management. Br J Haematol 168:639–645
Roach RE, Lijfering WM, van Hylckama VA, Helmerhorst FM, Rosendaal FR, Cannegieter SC (2013) The risk of venous thrombosis in individuals with a history of superficial vein thrombosis and acquired venous thrombotic risk factors. Blood 122(26):4264–4269
Musil D, Kaletova M, Herman J (2016) Risk factors for superficial vein thrombosis in patients with primary chronic venous disease. Vasa 45(1):63–66
van Langevelde K, Lijfering WM, Rosendaal FR, Cannegieter SC (2011) Increased risk of venous thrombosis in persons with clinically diagnosed superficial vein thrombosis: results from the MEGA study. Blood 118:4239–4241
Di Nisio M, Wichers IM, Middeldorp S (2013) Treatment for superficial Thrombophlebitis of the leg. Cochrane Database Syst Rev 4:CD004982
Prandoni P, Tormene D, Pesavento R (2005) High vs low doses of low-molecular-weight heparin for the treatment of superficial vein thrombosis of the legs: a double-blind, randomized trial. J Thromb Haemost 3:1152–1157
Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber S, Nelson M, Wells P, Gould M, Dentali F (2012) Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis: American College of Chest Physicians evidence based clinical practice guidelines. Chest 141:e419S-e494S
Becattini C, Agnelli G, Schenone A, Eichinger S, Bucherini E, Silingardi M, Bianchi M, Moia M, Ageno W, Vandelli MR, Grandone E, Prandoni P, WARFASA Investigators (2012) Aspirin for preventing the recurrence of venous thromboembolism. N Engl J Med 366(21):1959–1967
Agnelli G, Buller HR, Cohen A et al (2013) Apixaban for extended treatment of venous thromboembolism. N Engl J Med 368:699–708
Romualdi E, Donadini MP, Ageno W (2011) Oral rivaroxaban after symptomatic venous thromboembolism: the continued treatment study (EINSTEIN-extension study). Expert Rev Cardiovasc Ther 9:841–844
Schulman S, Kearon C, Kakkar AK et al (2013) Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med 368:709–718
Andreozzi GM, Bignamini AA, Davì G, Palareti G, Matuška J, Holý M, Pawlaczyk-Gabriel K, Džupina A, Sokurenko GY, Didenko YP, Andrei LD, Lessiani G, Visonà A, SURVET Study Investigators (2015) Sulodexide for the prevention of recurrent venous thromboembolism: the sulodexide in secondary prevention of recurrent deep vein thrombosis (SURVET) study: A multicenter, randomized, double-blind. Placebo-Controlled Trial. Circulation 132(20):1891–1897
Zanolo G, Giachetti C, Mascellani G, Magliocca R, Ferrari G (1984) Pharmacokinetic aspects of tritium-labeled glycosaminoglycans (mesoglycan), their absorption n rat and mokey, and tissue distribution in rat. Boll Chim farm 123:8–20
Prisma. Riassunto delle caratteristiche del prodotto (RCP). Last text revision: Jan 31, 2019
Ofosu FA, Modi GJ, Smith LM, Cerksus AL, Hirsh J, Blajchman MA (1984) Heparan sulfate and dermatan sulfate inhibit the generation of thrombin activity in plasma by complementary pathways. Blood 64:742–747
Tollefsen DM (1995) Insight into the mechanism of action of heparin cofactor II. Thromb Haemost 74:209–1214
Valvano A, Bosso G, Apuzzi V, Riccone F, Sacca L, Oliviero U (2015) Mesoglycan improves vascular reactivity and insulin sensitivity in patients with metabolic syndrome. Atherosclerosis 243:407–413
Allegra C, Antignani PL (2014) Quality of life as measured by the CIVIQ20 questionnaire following oral mesoglycan treatment of patients with chronic venous disease. Int Angiol 33:407–418
Tufano A, Arturo C, Cimino E, Di Minno MND, Di Capua M, Cerbone AM, and Di Minno G (2010) Mesoglycan: clinical evidences for use in vascular diseases, Int J Vas Med 2010:390643
Belvedere R, Morretta E, Pessolano E, et al. Mesoglycan exerts its fibrinolytic effect through the activation of annexin A2. J Cell Physiol 2020: 1–18
Derosa G, D’Angelo A, Romano D, Maffioli P (2017) Evaluation of the effects of mesoglycan on some markers of endothelial damage and walking distance in diabetic patients with peripheral arterial disease. Int J Mol Sci 18:572. https://doi.org/10.3390/ijms18030572
Vecchio F, Zanchin G, Maggioni F, Santambrogio C, De Zanche L (1993) Mesoglycan in treatment of patients with cerebral ischemia: effects on hemorheologic and hematochemical parameters. Acta Neurol 6:449–455
Andreozzi GM (2007) Efficacia del mesoglicano in pazienti con trombosi venosa e insufficienza venosa cronica. Minerva Cardioangiol 55(6):741–753
Prandoni P, Cattelan AM, Carta M (1989) Long-term sequelae of deep-vein thrombosis of the lower limbs. Experience with mesoglycan. Ann Ital Med Int 4:378–385
Rutherford RB, Padberg FT Jr, Comerota AJ, Kistner RL, Meissner M, Moneta GL (2000) American Venous Forum’s Ad Hoc Committee on Venous Outcomes Assessment Venous severity scoring: an adjunct to venous outcome assessment. J Vasc Surg 31:1307–1312
Lamping DL, Schroter S, Kurz X, Kahn SR, Abenhaim L (2003) Evaluating outcomes in chronic venous disorders of the leg: Development of a scientifically rigorous patient-reported measure of symptoms and quality of life. J Vasc Surg 37:410–419
Kakkos S, Rivera M, Matsagas M et al (2003) Validation of the new venous severity scoring system in varicose vein surgery. J Vasc Surg 38:224–228
Vasquez M, Rabe E, McLafferty R et al (2010) Revision of the venous clinical severity score: Venous outcomes consensus statement: Special communication of the American Venous Forum Ad Hoc Outcomes Working Group. J Vasc Surg 52:1387–1396
Kahn S, Lamping DL, Ducruet T (2006) VEINES-QOL/Sym questionnaire was a reliable and valid disease-specific quality of life measure for deep venous thrombosis. J Clin Epidemiol 59:1049–1056
Schulman S, Kearon C, Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis (2005) Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost 3(4):692–694
Camporese G, Bernardi E, Prandoni P et al (2008) Low-molecular-weight heparin versus graduated compression stockings for prevention of venous thromboembolism after knee arthroscopy. A randomised trial. Ann Intern Med 149:73–82
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG (2009) Research electronic data capture (REDCap) - a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42:377–381
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Consortia
Contributions
GC contributed to the study conception and design. Material preparation, data collection and analysis were performed by GC, FN, CB and EB. The first draft of the manuscript was written by GC and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Accepted as a poster presentation at ISTH 2023 Congress, June 24-28, Montreal; PB1490, presented on Tuesday June 27, 2023–6.30-7.30 pm.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Camporese, G., Bernardi, E., Bortoluzzi, C. et al. Mesoglycan for the secondary prevention of superficial vein thrombosis: a randomized, controlled, double-blind study (METRO Study)—rationale and protocol. J Thromb Thrombolysis 57, 226–234 (2024). https://doi.org/10.1007/s11239-023-02896-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-023-02896-6