
Abstract
Whether renal function is causally associated with venous thromboembolism (VTE) is not yet fully elucidated. We conducted a two-sample Mendelian randomization (MR) study to determine the causal effect of renal function, measured as estimated glomerular filtration rate (eGFR), on VTE. Single-nucleotide polymorphisms associated with eGFR were selected as instrumental variables at the genome-wide significance level (p < 5 × 10−8) from a meta-analysis of 122 genome-wide association studies including up to 1,046,070 individuals. Summary-level data for VTE were obtained from the FinnGen consortium (6913 VTE cases and 169,986 non-cases) and UK Biobank study (4620 VTE cases and 356,574 non-cases). MR estimates were calculated using the random-effects inverse-variance weighted method and combined using fixed-effects meta-analysis. Genetically predicted decreased eGFR was significantly associated with an increased risk of VTE in both FinnGen and UK Biobank. For one-unit decrease in log-transformed eGFR, the odds ratios of VTE were 2.93 (95% confidence interval (CI) 1.25, 6.84) and 4.46 (95% CI 1.59, 12.5) when using data from FinnGen and UK Biobank, respectively. The combined odds ratio was 3.47 (95% CI 1.80, 6.68). Results were consistent in all sensitivity analyses and no horizontal pleiotropy was detected. This MR-study supported a casual role of impaired renal function in VTE.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Highlights
-
Renal function has been associated with venous thromboembolism in observational studies.
-
The present Mendelian randomization study confirmed a causal association between impaired renal function and elevated risk of venous thromboembolism in two independent populations.
-
The findings highlight the importance of maintaining a healthy renal function in venous thromboembolism prevention.
Introduction
Chronic kidney disease is one of the major global public health issues, affecting 9.1% (corresponding to 697.5 million) of individuals and causing 35.8 million disability-adjusted life years worldwide in 2017 [1]. Impaired renal function reflected by low estimated glomerular filtration rate (eGFR) is associated with changes in certain coagulant and inflammatory factors [2, 3] and has been revealed to be associated with an increased risk of venous thromboembolism (VTE) in most observational studies [4,5,6,7]. The association has been seen not only in individuals with chronic kidney disease [6, 7] but also in those with normal eGFR [4]. However, inherited methodological limitations, such as residual confounding and reverse causality, in observational studies challenge the causal inference on the effect of renal function measured by eGFR on risk of VTE.
Utilizing genetic variants as instruments for an exposure (e.g., eGFR), the Mendelian randomization (MR) framework can strengthen the causal inference in an exposure-outcome association by reducing residual confounding and diminishing reverse causality [8, 9]. The rationale for the reduction in unobserved confounding is that genetic variants are randomly allocated at conception, and therefore, one trait is generally not correlated with other traits. This process resembles the random assignment of participants to treatment and control groups in a randomized controlled trial [8, 9]. The MR design also minimizes reverse causality because alleles are fixed at birth and cannot be modified by the onset or progression of the disease [8, 9]. Therefore, if a genetic variant that alters the exposed level or imitates its biological effects is also associated with the disease (but not stronger than exposure), this offers strong evidence that the exposure is a causal risk factor for the disease. Here, we conducted a two-sample MR analysis to assess whether a decline in eGFR is causally associated with an elevated risk of VTE.
Methods
Study design
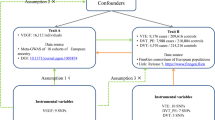
The study design overview is shown in Fig. 1. Genetic instruments for eGFR were obtained from a meta-analysis of 122 genome-wide association studies (GWASs) with up to 1,046,070 individuals. Summary-level genetic data for VTE were derived from the FinnGen consortium and UK Biobank study. Detailed information on used studies or consortium are displayed in Supplementary table 1. Original studies included in the meta-analysis of GWASs for eGFR, the FinnGen consortium, and the UK Biobank study have been approved by a relevant review board. The present analyses were approved by the Swedish Ethical Review Authority.

Study design overview. eGFR estimated glomerular filtration rate, SNPs single-nucleotide polymorphisms, VTE venous thromboembolism
Genetic instrument selection
Single-nucleotide polymorphisms associated with creatinine-based eGFR were extracted as instrumental variables at the genome-wide significance level (p < 5 × 10−8) from the discovery trans-ancestry meta-analysis of 121 GWASs including up to 765,348 individuals of multi-ancestries (around 74% European population) and a replication study including up to 280,772 European-descent participants (> 90% men and many with comorbidities such as hypertension [10]) from the Million Veteran Program [11]. The 308 SNPs, explaining around 7.1% of phenotypic variance, identified in the discovery stage were proposed as the primary genetic instruments for eGFR. The median age of participants in the discovery stage was 54 years and a half of them were women. After the exclusion of participants with extreme eGFR values (< 15 and > 200 ml/min per 1.73 m2), the median of the study-specific mean eGFR values was 89 ml/min per 1.73 m2 and the interquartile range was 81–94 ml/min per 1.73 m2. Three supplementary sets of instruments, including 264 eGFR-associated SNPs from both discovery and replication stages, 224 replicated SNPs associated with both eGFR (p < 5 × 10−8) and chronic kidney disease (p < 0.05), and 256 eGFR-associated SNPs derived from European populations, were utilized to assess the robustness of the primary genetic instrument. Genome-wide association test adjusted for age, sex, study site, genetic principal components, relatedness and other study-specific features. All used SNPs were independent and not in linkage disequilibrium with a distance over 500-kb flanking regions and r2 ≤ 0.01 in the 1000Gp1v3 dataset. Detailed information on the four sets of genetic instruments for eGFR is presented in Supplementary table 2.
VTE data sources
Summary genetic statistic data for VTE were available from the FinnGen consortium and UK Biobank study [12]. There was no participant overlap between the exposure and outcome populations. The FinnGen consortium (R4 release) includes 6913 VTE cases and 169,986 non-cases of Finnish descent. In FinnGen, individuals with ambiguous gender, high genotype missingness (> 5%), excess heterozygosity (± 4 standard deviation) and non-Finnish ancestry were excluded and the genetic variants with high missingness (> 2%), low Hardy–Weinberg equilibrium p-value (p < 5 × 10−6) and minor allele count, minor allele counts < 3 were excluded. Association tests were adjusted for age, sex, 10 genetic principal components and genotyping batch. Genotype imputation was done with the population specific SISu v3 reference panel. Detailed methods, including information on each included study, software used, fine-mapping and analytic codes, are presented in its website (https://www.finngen.fi/).
The data from the UK Biobank study were derived from Neale lab (the second wave, http://www.nealelab.is/uk-biobank) and encompasses 4620 VTE cases and 356,574 non-cases of British genetic ancestry. Individuals who are closely related (or at least one of a related pair of individuals) and those with sex chromosome aneuploidies were excluded. Neale lab restricted SNPs with minor allele frequency > 0.1% and Hardy–Weinberg equilibrium p-value > 1 × 10−10 and an info score > 0.8. The Haplotype Reference Consortium was used in imputation stage. There are around 54% female in all included participants. Association tests were adjusted for age, sex and up to 20 genetic principal components.
Statistical analysis
The random-effects inverse-variance weighted method was used for the main analysis. This method provides estimate with the highest precision and rely on the assumption that all SNPs are valid instrumental variables [13]. Results from inverse-variance weighted random-effects model based on the FinnGen consortium and UK Biobank study were combined using fixed-effect meta-analysis method. Four other MR methods, including inverse-variance weighted fixed-effects model, weighted median method [14], MR-Egger regression [15] and MR-PRESSO method [16], were employed as sensitivity analyses to examine the robustness of the results and correct for pleiotropy. The weighted median approach can provide consistent estimates if ≥ 50% of the weight in the analysis comes from valid instrumental variables [14]. The MR-Egger regression can detect and correct for directional pleiotropy albeit with compromised power [15]. The MR-PRESSO test can detect possible outliers and generate estimates after outliers removing, thereby correcting for horizontal pleiotropy [16]. The MR-PRESSO distortion test aims at assessing the differences between the estimates before and after outlier correction and a p < 0.05 of distortion test indicates a significant difference in estimates before and after outlier correction [16]. Funnel and scatter plots were generated to visualize the directional pleiotropy [15]. The I2 (%) statistic [17] and Cochrane’s Q value was calculated to assess the heterogeneity among estimates across individual SNPs. Odds ratios (ORs) and corresponding confidence intervals (CIs) of VTE were scaled to one-unit decrease in log-transformed eGFR. All analyses were performed using the mrrobust package [18] in Stata/SE 15.0 (Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC.) and the TwoSampleMR package [19] in R Software 3.6.0 (R Core Team. R Foundation for Statistical Computing. Vienna, Austria. 2019. https://www.R-project.org).
Results
All eGFR-associated SNPs were available in the UK Biobank. Twenty-three SNPs were missing in the FinnGen consortium dataset of which twenty-two were replaced with proxy SNPs with r2 > 0.8. The proxy SNPs used are shown in Supplementary table 3. F-statistics for four sets of genetic instruments were over 10 (Supplementary table 2). Funnel plots showed a symmetric distribution of SNPs in both analyses based on the FinnGen consortium and UK Biobank study (Supplementary Fig. 1).
Genetically predicted decreased eGFR was associated with an increased risk of VTE in both the FinnGen consortium and UK Biobank (Fig. 2). For one-unit increase in log-transformed eGFR, the ORs of VTE were 2.93 (95% CI 1.25, 6.84; p = 0.013) using data from FinnGen and 4.46 (95% CI 1.59, 12.5; p = 0.005) using data from UK Biobank. The combined OR was 3.47 (95% CI 1.80, 6.68; p < 0.001) in the meta-analysis. Results were consistent in all sensitivity analyses. Significant heterogeneity was detected across estimates from used SNPs in analyses based on data from both FinnGen (I2 = 16; Cochrane’s Q = 377; phet = 0.004) and UK Biobank (I2 = 25; Cochrane’s Q = 389; phet < 0.001). There was no horizontal pleiotropy in MR-Egger regression (p > 0.300). In the MR-PRESSO analyses, one and four outliers were detected using data from FinnGen and UK Biobank, respectively. However, p values for distortion tests were > 0.05, indicating no significant difference between estimates before and after outlier removal. Scatter plot of the association of eGFR with VTE using 308 SNPs is shown in Supplementary Fig. 2.

Association of genetically predicted eGFR with venous thromboembolism using 308 SNPs for eGFR. CI confidence interval, eGFR estimated glomerular filtration rate, IVW inverse-variance weighted, OR odds ratio, SNPs single-nucleotide polymorphisms. There were 10,023 venous thromboembolism cases and 486,809 non-cases in the meta-analysis of IVW-random effects model. Significant heterogeneity was detected among estimates from used SNPs in analyses based on data from both the FinnGen consortium (I2 = 19; Rucker’s Q = 377; phet = 0.004) and UK Biobank study (I2 = 25; Rucker’s Q = 389; phet < 0.001). There was no horizontal pleiotropy in MR-Egger regression using data from FinnGen consortium and UK Biobank study (p > 0.300). One and four outliers were detected in MR-PRESSO analyses using data from FinnGen consortium and UK Biobank study, respectively. However, p values for distortion tests were > 0.05, indicating no significant difference between estimates before and after outlier removing
Results of analyses using three supplementary sets of genetic instrumental variables for eGFR are presented in Table 1. Overall, the associations of genetically predicted eGFR with VTE risk persisted in all analyses based on the inverse-variance weighted method albeit with significant heterogeneity among used SNPs (p < 0.05). We did not detect horizontal pleiotropy in all MR-Egger regression (p > 0.05). One to six outliers were observed in MR-PRESSO analyses. However, all p values for distortion tests were > 0.05.
Discussion
The present two-sample MR study strengthened the evidence of a causal association between eGFR and VTE (Figs. 2, 3). The association was consistent in two independent populations and remained stable using different sets of instrumental variables for eGFR and in all statistical models.

Summarizing figure on MR association between eGFR and VTE risk. eGFR estimated glomerular filtration rate, MR Mendelian randomization, VTE venous thromboembolism
Our finding is in line with observational studies [4,5,6,7]. The Longitudinal Investigation of Thromboembolism Etiology study including 19,073 middle-aged and elderly adults found that individuals with stage 3/4 chronic kidney disease (eGFR between 15 and 59 ml/min per 1.73 m2) had 28% higher risk of VTE compared with those with normal renal function in a follow-up period of 11.8-year [7]. A large pooled individual participant data additionally revealed a dose–response relationship between eGFR and VTE risk and specified that eGFR could be an independent predictor for VTE risk even among individual with normal eGFR range [4]. The present study used MR design empowered the causal inference in the association between eGFR and VTE and confirmed the protective effect of high eGFR on VTE.
There are several potential biological pathways underlying the link between eGFR and VTE [2, 20]. In particular, pre-coagulation is activated in patients with insufficient renal function, and this may accelerate the development of VTE. Impaired kidney function is associated with elevated levels of coagulation factors, such as D-dimer, fibrinogen, factor VII, and factor VIII and von Willebrand factor [2, 3, 21, 22]. On the other hand, endogenous anticoagulants, such as antithrombin, are decreased among individuals with impaired kidney function due to increased urinary loss of antithrombin out of proportion to synthesis [23]. Change in antithrombin, however, was not observed in one case–control study [3] and the alternation of anticoagulants might occur in severe chronic kidney disease mainly. Hypoalbuminemia caused by kidney disease (nephrotic syndrome) results in increased availability of thromboxane A2 that enhances platelet activation and aggregation, thereby facilitating thrombus formation [24]. In addition, some studies indicated that decreased activity of the fibrinolytic system might also mediate the association between renal function and VTE. However, data on this pathway are inconclusive and needs to be unified [25, 26].
There are several strengths and limitations of the present study. The major merit is the MR design which greatly strengthened the causal inference on the associations between eGFR and VTE by reducing residual confounding and reverse causality. Additionally, the association was tested and replicated in two independent data sources. The high consistency between findings from FinnGen consortium and UK Biobank study gave a boost to the possibility that the finding is causal. A limitation is that there might be population bias in the finding using genetic instruments derived from population of multi-ancestries. However, a high proportion of European population (around 74%) and a consistent result based on merely European population minimized the possibility that our finding was affected by population bias. The GWAS meta-analysis on eGFR found that eGFR was genetically correlated with lean mass and physical fitness (rg = − 0.20), which might be a pleiotropic factor influencing the established association between eGFR and VTE. However, a null association between lean mass and VTE in a recent MR study [27] and no directional pleiotropy detected in the present MR-Egger regression analysis indicated a negligible distortion by potential pleiotropy. Chronic kidney disease and VTE are both frequently observed in cancer patients [28, 29]. We cannot rule out that the eGFR-associated SNPs have pleiotropic associations with cancer, and that the observed association between eGFR and VTE is to some extent influenced by cancer. In addition, as we used summary-level data, we were unable to assess the dose–response relationship between eGFR and VTE and could not stratify the analyses by impaired renal function.
Conclusions
The present study provides the first MR evidence that declined renal function, measured as decreased eGFR, is causally associated with an increased risk of VTE. The finding suggests that clinicians need to optimize medical treatment to maintain healthy renal function of patients to prevent future VTE and VTE screening and prophylaxis should be reinforced among individuals with impaired renal function.
Data availability
The datasets analyzed in this study are publicly available summary statistics. Data used in the present study are available in OSF data respiratory (https://osf.io/bk52p/).
References
GBD Chronic Kidney Disease Collaboration (2020) Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395(10225):709–733
Cheung KL, Zakai NA, Callas PW, Howard G, Mahmoodi BK, Peralta CA, Judd SE, Kurella Tamura M, Cushman M (2018) Mechanisms and mitigating factors for venous thromboembolism in chronic kidney disease: the REGARDS study. J Thromb Haemost 16(9):1743–1752
Ocak G, Vossen CY, Lijfering WM, Verduijn M, Dekker FW, Rosendaal FR, Cannegieter SC (2014) Role of hemostatic factors on the risk of venous thrombosis in people with impaired kidney function. Circulation 129(6):683–691
Mahmoodi BK, Gansevoort RT, Næss IA, Lutsey PL, Brækkan SK, Veeger NJ, Brodin EE, Meijer K, Sang Y, Matsushita K, Hallan SI, Hammerstrøm J, Cannegieter SC, Astor BC, Coresh J, Folsom AR, Hansen JB, Cushman M (2012) Association of mild to moderate chronic kidney disease with venous thromboembolism: pooled analysis of five prospective general population cohorts. Circulation 126(16):1964–1971
Parikh AM, Spencer FA, Lessard D, Emery C, Baylin A, Linkletter C, Goldberg RJ (2011) Venous thromboembolism in patients with reduced estimated GFR: a population-based perspective. Am J Kidney Dis 58(5):746–755
Cheung KL, Zakai NA, Folsom AR, Kurella Tamura M, Peralta CA, Judd SE, Callas PW, Cushman M (2017) Measures of kidney disease and the risk of venous thromboembolism in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) Study. Am J Kidney Dis 70(2):182–190
Wattanakit K, Cushman M, Stehman-Breen C, Heckbert SR, Folsom AR (2008) Chronic kidney disease increases risk for venous thromboembolism. J Am Soc Nephrol 19(1):135–140
Burgess S, Thompson SG (2015) Mendelian randomization: methods for using genetic variants in causal estimation. Chapman & Hall/CRC Press, London
Smith GD, Ebrahim S (2003) “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32(1):1–22
Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, Guarino P, Aslan M, Anderson D, LaFleur R, Hammond T, Schaa K, Moser J, Huang G, Muralidhar S, Przygodzki R, O’Leary TJ (2016) Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 70:214–223
Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, Tin A, Wang L, Chu AY, Hoppmann A, Kirsten H, Giri A, Chai JF, Sveinbjornsson G, Tayo BO, Nutile T, Fuchsberger C, Marten J, Cocca M, Ghasemi S, Xu Y, Horn K, Noce D, van der Most PJ, Sedaghat S, Yu Z, Akiyama M, Afaq S, Ahluwalia TS, Almgren P, Amin N, Ärnlöv J, Bakker SJL, Bansal N, Baptista D, Bergmann S, Biggs ML, Biino G, Boehnke M, Boerwinkle E, Boissel M, Bottinger EP, Boutin TS, Brenner H, Brumat M, Burkhardt R, Butterworth AS, Campana E, Campbell A, Campbell H, Canouil M, Carroll RJ, Catamo E, Chambers JC, Chee ML, Chee ML, Chen X, Cheng CY, Cheng Y, Christensen K, Cifkova R, Ciullo M, Concas MP, Cook JP, Coresh J, Corre T, Sala CF, Cusi D, Danesh J, Daw EW, de Borst MH, De Grandi A, de Mutsert R, de Vries APJ, Degenhardt F, Delgado G, Demirkan A, Di Angelantonio E, Dittrich K, Divers J, Dorajoo R, Eckardt KU, Ehret G, Elliott P, Endlich K, Evans MK, Felix JF, Foo VHX, Franco OH, Franke A, Freedman BI, Freitag-Wolf S, Friedlander Y, Froguel P, Gansevoort RT, Gao H, Gasparini P, Gaziano JM, Giedraitis V, Gieger C, Girotto G, Giulianini F, Gögele M, Gordon SD, Gudbjartsson DF, Gudnason V, Haller T, Hamet P, Harris TB, Hartman CA, Hayward C, Hellwege JN, Heng CK, Hicks AA, Hofer E, Huang W, Hutri-Kähönen N, Hwang SJ, Ikram MA, Indridason OS, Ingelsson E, Ising M, Jaddoe VWV, Jakobsdottir J, Jonas JB, Joshi PK, Josyula NS, Jung B, Kähönen M, Kamatani Y, Kammerer CM, Kanai M, Kastarinen M, Kerr SM, Khor CC, Kiess W, Kleber ME, Koenig W, Kooner JS, Körner A, Kovacs P, Kraja AT, Krajcoviechova A, Kramer H, Krämer BK, Kronenberg F, Kubo M, Kühnel B, Kuokkanen M, Kuusisto J, La Bianca M, Laakso M, Lange LA, Langefeld CD, Lee JJ, Lehne B, Lehtimäki T, Lieb W; Lifelines Cohort Study, Lim SC, Lind L, Lindgren CM, Liu J, Liu J, Loeffler M, Loos RJF, Lucae S, Lukas MA, Lyytikäinen LP, Mägi R, Magnusson PKE, Mahajan A, Martin NG, Martins J, März W, Mascalzoni D, Matsuda K, Meisinger C, Meitinger T, Melander O, Metspalu A, Mikaelsdottir EK, Milaneschi Y, Miliku K, Mishra PP; V. A. Million Veteran Program, Mohlke KL, Mononen N, Montgomery GW, Mook-Kanamori DO, Mychaleckyj JC, Nadkarni GN, Nalls MA, Nauck M, Nikus K, Ning B, Nolte IM, Noordam R, Oconnell J, Odonoghue ML, Olafsson I, Oldehinkel AJ, Orho-Melander M, Ouwehand WH, Padmanabhan S, Palmer ND, Palsson R, Penninx BWJH, Perls T, Perola M, Pirastu M, Pirastu N, Pistis G, Podgornaia AI, Polasek O, Ponte B, Porteous DJ, Poulain T, Pramstaller PP, Preuss MH, Prins BP, Province MA, Rabelink TJ, Raffield LM, Raitakari OT, Reilly DF, Rettig R, Rheinberger M, Rice KM, Ridker PM, Rivadeneira F, Rizzi F, Roberts DJ, Robino A, Rossing P, Rudan I, Rueedi R, Ruggiero D, Ryan KA, Saba Y, Sabanayagam C, Salomaa V, Salvi E, Saum KU, Schmidt H, Schmidt R, Schöttker B, Schulz CA, Schupf N, Shaffer CM, Shi Y, Smith AV, Smith BH, Soranzo N, Spracklen CN, Strauch K, Stringham HM, Stumvoll M, Svensson PO, Szymczak S, Tai ES, Tajuddin SM, Tan NYQ, Taylor KD, Teren A, Tham YC, Thiery J, Thio CHL, Thomsen H, Thorleifsson G, Toniolo D, Tönjes A, Tremblay J, Tzoulaki I, Uitterlinden AG, Vaccargiu S, van Dam RM, van der Harst P, van Duijn CM, Velez Edward DR, Verweij N, Vogelezang S, Völker U, Vollenweider P, Waeber G, Waldenberger M, Wallentin L, Wang YX, Wang C, Waterworth DM, Bin Wei W, White H, Whitfield JB, Wild SH, Wilson JF, Wojczynski MK, Wong C, Wong TY, Xu L, Yang Q, Yasuda M, Yerges-Armstrong LM, Zhang W, Zonderman AB, Rotter JI, Bochud M, Psaty BM, Vitart V, Wilson JG, Dehghan A, Parsa A, Chasman DI, Ho K, Morris AP, Devuyst O, Akilesh S, Pendergrass SA, Sim X, Böger CA, Okada Y, Edwards TL, Snieder H, Stefansson K, Hung AM, Heid IM, Scholz M, Teumer A, Köttgen A, Pattaro C (2019) A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet 51(6): 957–972
Sudlow C, Gallacher J, Allen N et al (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12(3):e1001779. https://doi.org/10.1371/journal.pmed.1001779
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG (2017) Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology 28(1):30–42. https://doi.org/10.1097/ede.0000000000000559
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40(4):304–314. https://doi.org/10.1002/gepi.21965
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44(2):512–525. https://doi.org/10.1093/ije/dyv080
Verbanck M, Chen CY, Neale B, Do R (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 50(5):693–698
Higgins JP, Thompson SG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21(11):1539–1558
Spiller W, Davies NM, Palmer TM (2019) Software application profile: mrrobust—a tool for performing two-sample summary Mendelian randomization analyses. Int J Epidemiol 48(3):6
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR, Haycock PC (2018) The MR-Base platform supports systematic causal inference across the human phenome. Elife 7:e34408
Wattanakit K, Cushman M (2009) Chronic kidney disease and venous thromboembolism: epidemiology and mechanisms. Curr Opin Pulm Med 15(5):408–412
Keller C, Katz R, Cushman M, Fried LF, Shlipak M (2008) Association of kidney function with inflammatory and procoagulant markers in a diverse cohort: a cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis (MESA). BMC Nephrol 9:9
Shlipak MG, Fried LF, Crump C, Bleyer AJ, Manolio TA, Tracy RP, Furberg CD, Psaty BM (2003) Elevations of inflammatory and procoagulant biomarkers in elderly persons with renal insufficiency. Circulation 107(1):87–92
Mahmoodi BK, ten Kate MK, Waanders F, Veeger NJ, Brouwer JL, Vogt L, Navis G, van der Meer J (2008) High absolute risks and predictors of venous and arterial thromboembolic events in patients with nephrotic syndrome: results from a large retrospective cohort study. Circulation 117(2):224–230
Jackson CA, Greaves M, Patterson AD, Brown CB, Preston FE (1982) Relationship between platelet aggregation, thromboxane synthesis and albumin concentration in nephrotic syndrome. Br J Haematol 52(1):69–77
Folsom AR, Cushman M, Heckbert SR, Rosamond WD, Aleksic N (2003) Prospective study of fibrinolytic markers and venous thromboembolism. J Clin Epidemiol 56(6):598–603
Lisman T, de Groot PG, Meijers JC, Rosendaal FR (2005) Reduced plasma fibrinolytic potential is a risk factor for venous thrombosis. Blood 105(3):1102–1105
Larsson SC, Bäck M, Rees JMB, Mason AM, Burgess S (2020) Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: a Mendelian randomization study. Eur Heart J 41(2):221–226
Torres da Costa ESV, Costalonga EC, Coelho FO, Caires RA, Burdmann EA (2018) Assessment of kidney function in patients with cancer. Adv Chronic Kidney Dis 25(1):49–56
Ay C, Pabinger I, Cohen AT (2017) Cancer-associated venous thromboembolism: burden, mechanisms, and management. Thromb Haemost 117(2):219–230
Acknowledgements
Genetic instruments for estimated glomerular filtration rate were obtained from a published meta-analysis of genome-wide association studies (Wuttke M et al. 2019; PubMed identifier 31152163). Summary-level data for venous thromboembolism were obtained from the FinnGen consortium (https://www.finngen.fi/) and UK Biobank study (Neale lab; http://www.nealelab.is/uk-biobank). The authors thank all investigators for sharing these data.
Funding
Open access funding provided by Karolinska Institute. This work was funded by the Karolinska Institutet’s Research Foundation Grants (Grant Number 2020-01842), the Swedish Research Council for Health, Working Life and Welfare (Forte; Grant No. 2018-00123), the Swedish Research Council (Vetenskapsrådet; Grant No. 2019-00977), and the Swedish Heart–Lung Foundation (Hjärt-Lungfonden; Grant No. 20190247). Maria Bruzelius is funded by Stockholm County Council (Grant No. K2892-2016).
Author information
Authors and Affiliations
Contributions
SY drafted the manuscript. SY and SCL collected and analyzed the data. SY, MB and SCL designed the study, interpreted the data and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, S., Bruzelius, M. & Larsson, S.C. Causal effect of renal function on venous thromboembolism: a two-sample Mendelian randomization investigation. J Thromb Thrombolysis 53, 43–50 (2022). https://doi.org/10.1007/s11239-021-02494-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-021-02494-4