
Abstract
Apixaban (BMS-562247; 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide), a direct inhibitor of activated factor X (FXa), is in development for the prevention and treatment of various thromboembolic diseases. With an inhibitory constant of 0.08 nM for human FXa, apixaban has greater than 30,000-fold selectivity for FXa over other human coagulation proteases. It produces a rapid onset of inhibition of FXa with association rate constant of 20 μM−1/s approximately and inhibits free as well as prothrombinase- and clot-bound FXa activity in vitro. Apixaban also inhibits FXa from rabbits, rats and dogs, an activity which parallels its antithrombotic potency in these species. Although apixaban has no direct effects on platelet aggregation, it indirectly inhibits this process by reducing thrombin generation. Pre-clinical studies of apixaban in animal models have demonstrated dose-dependent antithrombotic efficacy at doses that preserved hemostasis. Apixaban improves pre-clinical antithrombotic activity, without excessive increases in bleeding times, when added on top of aspirin or aspirin plus clopidogrel at their clinically relevant doses. Apixaban has good bioavailability, low clearance and a small volume of distribution in animals and humans, and a low potential for drug–drug interactions. Elimination pathways for apixaban include renal excretion, metabolism and biliary/intestinal excretion. Although a sulfate conjugate of Ο-demethyl apixaban (O-demethyl apixaban sulfate) has been identified as the major circulating metabolite of apixaban in humans, it is inactive against human FXa. Together, these non-clinical findings have established the favorable pharmacological profile of apixaban, and support the potential use of apixaban in the clinic for the prevention and treatment of various thromboembolic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombosis is a major cause of morbidity and mortality in the Western world and plays a pivotal role in the pathogenesis of numerous cardiovascular disorders, including acute coronary syndrome (ACS) (i.e. unstable angina and myocardial infarction), sudden cardiac death, peripheral arterial occlusion, ischemic stroke, deep vein thrombosis (DVT) and pulmonary embolism. Despite recent advances in interventional and drug therapy for thrombosis, the burden of thrombotic disease remains unacceptably high [1, 2]. There is therefore a significant need for new antithrombotic therapies that are more effective and provide improved safety profile compared with current treatments. This review focuses on the pre-clinical discovery of apixaban, a promising new oral antithrombotic agent that specifically targets activated factor X (FXa) of the blood coagulation cascade.
Drug discovery strategy—targeting factor Xa
As the last serine protease in the blood coagulation cascade, thrombin is the key enzyme responsible for physiological fibrin clot formation and platelet activation. Thrombin also plays a prominent role in the pathologic generation of occlusive thrombi in arteries or veins, a process that may lead to arterial or venous thrombotic disease. Thus, attenuation of the activity of thrombin—either via direct inhibition or via blockade of other proteases that lie upstream in the coagulation cascade and are intimately involved in thrombin generation (e.g. FXa)—has been intensively investigated as a novel means to prevent and treat thrombotic disease.
Three key observations supported our hypothesis that inhibition of FXa may represent an acceptable approach for effective and safe antithrombotic therapy. First, as the process of blood coagulation involves sequential activation and amplification of coagulation proteins, generation of one molecule of FXa can lead to the activation of hundreds of thrombin molecules [3]. In principle, therefore, inhibition of FXa may represent a more efficient way of reducing fibrin clot formation than direct inhibition of thrombin activity. This principle is consistent with an in vitro observation, suggesting that inhibition of FXa but not thrombin may result in a more effective sustained reduction of thrombus-associated procoagulant activity [4]. Second, inhibition of FXa is not thought to affect existing levels of thrombin. Further, reversible FXa inhibitors might not completely suppress the production of thrombin. These small amounts of thrombin might be sufficient to activate high affinity platelet thrombin receptors to permit physiological regulation of hemostasis. Indeed, experimental evidence from animal studies suggests that the antithrombotic efficacy of FXa inhibitors is accompanied by a lower risk of bleeding when compared with thrombin inhibitors [5–7] (for review, see Hauptmann and Stürzebecher [8] and Leadley [9]). Finally, the strongest evidence for FXa as an antithrombotic drug target is the clinical proof of concept studies of the indirect FXa inhibitor fondaparinux [10]. Taken together, these observations suggest that inhibition of FXa is a potentially attractive antithrombotic strategy.
We initiated a drug discovery program on small-molecule direct FXa inhibitors, with the goal of identifying novel oral anticoagulants not burdened by the well-known limitations of vitamin K antagonists such as warfarin, agents that remain the only oral anticoagulants approved for long-term use until very recently [11]. (On October 19, 2010, FDA approved the oral direct thrombin inhibitor dabigatran etexilate to prevent stroke and blood clots in patients with non-valvular atrial fibrillation [12].) These new FXa inhibitors would have the following target profile. First, they would be direct, highly selective and reversible inhibitors of FXa, with a rapid onset of action, and would demonstrate a relatively wide therapeutic index and few food and drug interactions (thereby avoiding the need for frequent coagulation monitoring and dose adjustment). Second, these FXa inhibitors would have predictable pharmacokinetic and pharmacodynamic profiles that allow fixed oral dosing, accompanied by low peak-to-trough plasma concentrations that provide high levels of efficacy and low rates of bleeding. Finally, as the FXa target resides in the central or blood compartment, the pharmacokinetic profile of these agents would also feature a low volume of distribution (to minimize off-target risks) and low systemic clearance (to reduce the potential for drug-drug interactions).
Based on many years of research and development, we have identified the potent, highly selective and direct FXa inhibitor, apixaban (BMS-562247) [13–15]. Apixaban is one of the most promising specific, single-target oral anticoagulants in late clinical development. In clinical trials, apixaban has been shown to provide predictable and consistent anticoagulation, accompanied by promising efficacy and safety profiles in the prevention and treatment of various thromboembolic diseases [16–22]. The pharmacological and clinical profiles of apixaban suggest that it has the potential to address many of the limitations of warfarin therapy, currently the standard of care in chronic oral anticoagulation. In this review, we summarize the chemistry and pre-clinical profile of apixaban.
Chemistry
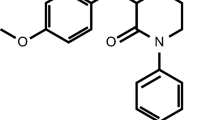
Apixaban is a small-molecule, selective FXa inhibitor. It is chemically described as 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide. The molecular formula for apixaban is C25H25N5O4, which corresponds to a molecular weight of 459.5.
Discovery of apixaban
In the early 1990s, DuPont scientists invested a great amount of effort in the development of inhibitors of glycoprotein IIb/IIIa. These efforts resulted in several compounds that were advanced to clinical trials as potential anti-platelet agents. By the mid-1990s, scientists at DuPont had recognized similarities between the platelet glycoprotein GPIIb/IIIa peptide sequence Arg-Gly-Asp (RGD) and the prothrombin substrate FXa sequence, Glu-Gly-Arg (EGR). Consequently, a high-throughput lead evaluation program was initiated to screen the IIb/IIIa library for FXa inhibitory activity. This effort resulted in the identification of a small number of isoxazoline derivatives such as 1 (FXa K i = 38.5 μM) (Fig. 1) [23]. Using molecular modeling and structure-based design, an optimization strategy resulted in the identification of a benzamidine containing FXa inhibitor 2 (SF303) with enhanced potency (FXa K i = 6.3 nM) and potent antithrombotic activity in an experimental model of thrombosis [24–26]. Aside from the key amidine P1 and the enzyme Asp189 interaction, the biarylsulfonamide P4 moiety was designed to neatly stack in the S4 hydrophobic box of FXa, which contains the residues Tyr99, Phe174 and Trp215, with the terminal O-phenylsulfonamide ring making an edge-to-face interaction with Trp215. Subsequent re-optimizations led to vicinally substituted isoxazole analogs such as compound 3, which retained anti-FXa potency (FXa K i = 0.1 nM) [27] and a pyrazole analog 4 (SN429), which demonstrated 13 pM binding affinity against FXa and good antithrombotic activity in a rabbit model of thrombosis [28, 29]. The discovery of SN429 was tremendously important in that it set the stage for an optimization strategy that led to the discovery of several important compounds, such as 5 (DPC423), a phase I clinical candidate with a long terminal half-life of approximately 30 h in humans [6, 28–30], and 6 (razaxaban) [31, 32], a compound that was advanced to a phase II proof-of-principle clinical trial. In fact, razaxaban was the first small molecule FXa inhibitor to provide clinical validation of the effectiveness of FXa inhibition strategies [33].

The evolution of the pyrazole-based FXa inhibitors: the discovery of apixaban
Development of razaxaban was quickly followed by the identification of a novel bicyclic tetrahydropyrazolo-pyridinone analog 7 (BMS-740808, FXa K i = 0.03 nM) [34]. The evolution of the bicyclic pyrazole template allowed for the incorporation of a diverse set of P1 groups, the most important of which was the p-methoxyphenyl analog 8 (K i = 0.14 nM) [13]. Compound 8 retained potent FXa affinity and good anticoagulant activity in vitro, was efficacious in in vivo rabbit antithrombotic models and showed high oral bioavailability in dogs. A significant breakthrough was subsequently achieved, via the incorporation of a pendent P4 lactam group and a carboxamido pyrazole moiety, that led to the discovery of 9 (BMS-562247, FXa K i = 0.08 nM) [13], a highly potent and selective FXa inhibitor with good efficacy in various animal models of thrombosis. Importantly, compound 9 also showed an excellent pharmacokinetic profile in dogs, with low clearance, low volume of distribution and high oral bioavailability [13]. The superior pre-clinical profile demonstrated by 9 enabled its rapid progression into clinical development as apixaban [15]. Figure 2 illustrates the X-ray structure of apixaban bound to FXa and shows the p-methoxyphenyl P1 deeply inserted into the S1 pocket, with the aryllactam P4 moiety neatly stacked in the hydrophobic S4 pocket.

X-ray structure of apixaban bound to factor Xa
In vitro pharmacology
Potency, selectivity and kinetic mode of inhibition
Apixaban is a highly potent, reversible, active-site inhibitor of human FXa, with a K i of 0.08 nM at 25°C and 0.25 nM at 37°C in the FXa tripeptide substrate (N-α-benzyloxycarbonyl-d-Arg-Gly-Arg-pNA) assay [35]. Analysis of enzyme kinetics shows that apixaban acts as a competitive inhibitor of FXa versus the synthetic tripeptide substrate, indicating that it binds in the active site. Apixaban produces a rapid onset of inhibition under a variety of conditions with association rate constant of 20 (μM−1/s approximately, and shows competitive inhibition of FXa versus the synthetic tripeptide substrate. Reversibility of FXa inhibition is demonstrated by the recovery of FXa activity at 37°C upon 200-fold dilution of a pre-formed FXa:apixaban complex into tripeptide substrate, an effect associated with a dissociation rate constant of ~0.0113 s−1. Unlike indirect inhibitors of thrombin and FXa, such as heparin, the low molecular weight heparins and fondaparinux, apixaban, a direct FXa inhibitor, does not require the presence of antithrombin III to inhibit FXa. As shown in Table 1, apixaban has greater than 30,000-fold selectivity for FXa relative to other human coagulation proteases and structurally related enzymes involved in digestion and fibrinolysis [13].
In the prothrombinase assay, apixaban is an effective inhibitor of the action of human FXa on its physiological substrate, prothrombin, blocking the action of FXa on prothrombin within the prothrombinase complex with a K i of 0.62 nM [35]. It should be noted that when apixaban was evaluated as an inhibitor of FXa versus the physiological substrate prothrombin in its prothrombinase state, non-competitive inhibition was observed. This finding is consistent with prothrombin binding being dictated primarily by interactions at exosites of FXa [36]. Apixaban also inhibits thrombus-associated FXa activity with a concentration causing 50% inhibition (IC50) of 1.3 nM [37]. In summary, apixaban is capable of inhibiting the activity of free FXa, thrombus-associated FXa and FXa within the prothrombinase complex. Apixaban is a direct inhibitor of FXa from rats, rabbits and dogs, with K i values of 1.3, 0.16 and 1.7 nM, respectively (Table 2 [15]). Previous studies involving other small molecule, direct FXa inhibitors have also reported a species difference in FXa inhibition among humans, rabbits, rats and dogs [29, 38, 39].
In vitro pharmacodynamic studies
To evaluate the in vitro pharmacodynamic activity of apixaban in human plasma, studies were undertaken to examine [1] thrombin generation, [2] anticoagulant activity and [3] platelet aggregation. By inhibiting FXa, apixaban prevents the conversion of prothrombin to thrombin, resulting in decreased generation of thrombin. Using the thrombogram method, apixaban was shown to inhibit tissue factor-initiated thrombin generation in human platelet-poor plasma in vitro. The IC50 of the rate of thrombin generation was 50 nM, and the IC50 for attenuation of the peak thrombin concentration was 100 nM [40]. In human platelet-rich plasma, apixaban inhibited tissue factor-induced thrombin generation, as measured by the release of prothrombin fragment 1 + 2, with an IC50 of 37 nM [41].
As expected for an inhibitor of FXa, addition of apixaban to normal human plasma prolonged clotting times, including activated partial thromboplastin time (aPTT), prothrombin time (PT), modified PT (mPT, using diluted PT reagent) and HepTest. Among the three clotting time assays, it appears that the mPT and HepTest are 10–20 times more sensitive than aPTT and PT in monitoring the in vitro anticoagulant effect of apixaban in human plasma (Table 2 [15]). In both the PT and aPTT assays, apixaban had the highest potency in human and rabbit plasma, but was less potent in rat and dog plasma, which parallels its inhibitory potencies (K i) against human, rabbit, rat and dog FXa (Table 2 [15]).
In the human platelet aggregation assay, apixaban had no direct effects on platelet aggregation response to ADP, collagen, γ-thrombin, α-thrombin and TRAP [15, 41]. However, it indirectly inhibited platelet aggregation induced by thrombin derived from tissue factor-mediated coagulation pathway, with an IC50 of 4 nM [41]. The potent indirect antiplatelet effect of apixaban, together with its direct antithrombotic and anticoagulant activity, suggests that apixaban may possess dual mechanisms to prevent and treat both venous (platelet-poor and fibrin-rich) and arterial (platelet-rich and fibrin-poor) thrombosis. It should be noted that the in vitro tissue factor model of platelet aggregation is a useful tool for evaluation of the antiplatelet mechanisms of action of anticoagulants. However, caution should be exercised as in vitro antiplatelet potencies of compounds obtained in this model may not directly translate into antithrombotic potencies in patients in whom multiple prothrombotic mechanisms, complications of cardiovascular disease and polypharmacy are common.
In vivo pharmacology
The non-clinical pharmacology of apixaban has been studied in vivo in rats and rabbits. Its in vivo effects were assessed over a comprehensive dose range in various well-established non-clinical models of thrombosis and hemostasis. These non-clinical models have been well characterized with standard antiplatelet agents and anticoagulants, making them appropriate for evaluating the antithrombotic potential and bleeding liability of apixaban.
Antithrombotic and bleeding time effects in rats
Dose-dependent effects of apixaban were examined in a broad range of experimental models of thrombosis and hemostasis in rats [42]. Efficacy was evaluated using established models of thrombosis, including arterial-venous shunt thrombosis (AV-ST), tissue factor-stasis venous thrombosis, and FeCl2-induced vena cava thrombosis and carotid artery thrombosis. Hemostasis was assessed in models of cuticle bleeding time, renal cortex bleeding time and mesenteric bleeding time. Apixaban was given by a continuous intravenous (IV) infusion 1 h prior to the induction of thrombosis or bleeding.
Apixaban at 0.1, 0.3, 1 and 3 mg/kg/h IV produced dose-dependent increases in ex vivo PT (1.24, 1.93, 2.75 and 3.98 times control, respectively). In the various models of thrombosis, doses and plasma concentrations of apixaban for 50% thrombus reduction ranged from 0.39 to 1.55 mg/kg/h and 1.84 to 7.57 μM, respectively (Table 3) [42]. The 3 mg/kg/h dose of apixaban increased cuticle, renal and mesenteric bleeding times to 1.92, 2.13 and 2.98 times control, respectively. Bleeding time was not increased by apixaban at 0.1 and 0.3 mg/kg/h in any model. The 1 mg/kg/h dose produced an increase in mesenteric bleeding time, but showed no effect on renal or cuticle bleeding time. In comparison, heparin increased renal and cuticle bleeding times to two times those of apixaban when given at a dose (300 U/kg plus 10 U/kg/min IV) that matched the efficacy of apixaban (3 mg/kg/h IV) in arterial thrombosis. These studies demonstrate that in rats, apixaban has broad-spectrum antithrombotic efficacy and that these beneficial effects can be obtained at doses that show limited activity in multiple models of provoked bleeding.
Antithrombotic and bleeding time effects in rabbits
The antithrombotic efficacy of apixaban was evaluated in anesthetized rabbits using established models of thrombosis, including AV-ST, electrically induced carotid arterial thrombosis (ECAT) and DVT (a thread-induced vena cava thrombosis model). Hemostasis was assessed in a rabbit model of cuticle bleeding time. Apixaban was given by a continuous IV infusion 1 h prior to the induction of thrombosis or cuticle incision.
Antithrombotic studies
Apixaban exhibited strong antithrombotic activity in the rabbit models of AV-ST, ECAT and DVT, which compared well with standard antithrombotic agents (Figs. 3, 4) [7, 15, 43]. For instance, apixaban, the direct FXa inhibitor rivaroxaban, the direct thrombin inhibitor dabigatran and the oral anticoagulant warfarin showed similar efficacy in the prevention model of DVT (Fig. 3) [7, 15]. In the prevention model of ECAT, apixaban was as efficacious as the antiplatelet agent clopidogrel and warfarin (Fig. 4) [7, 15, 43]. Doses and plasma concentrations of apixaban for 50% thrombus reduction ranged from 0.07 to 0.27 mg/kg/h and 0.065 to 0.36 μM, respectively (Table 3) [15]. The 1 mg/kg/h dose was associated with approximately 80% antithrombotic efficacy in these models. Interestingly, the potency of apixaban in arterial and venous thrombosis prevention models was broadly equivalent. Apixaban also effectively inhibited the growth of a pre-formed intravascular thrombus in a treatment model of DVT, suggesting that apixaban shows potential for the treatment of established thrombosis [7].

Plots of thrombus reduction and bleeding time versus dose in apixaban, rivaroxaban, dabigatran and warfarin-treated rabbits. Thrombus reduction, measured in the prevention model of venous thrombosis, was expressed as the percentage reduction in thrombus weight after treatment, relative to the mean vehicle thrombus weight. Bleeding time effect was expressed as a ratio of treated versus the mean vehicle value. Data are mean ± SE (n = 6 per group for the thrombosis and bleeding time studies and n = 12 per dose for plasma concentrations) (data from Wong et al. [7, 15]; reproduced with permission). Reproduced from “Favorable therapeutic index of the direct factor Xa inhibitors, apixaban and rivaroxaban, compared with the thrombin inhibitor dabigatran in rabbits” published in “Journal of Thrombosis and Haemostasis” (2009), John Wiley and Sons; and from "Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies" published in “Journal of Thrombosis and Haemostasis” (2008), John Wiley and Sons

Dose-dependent effects of apixaban, clopidogrel and warfarin on integrated blood flow in the electrically induced carotid arterial thrombosis rabbit model. Data are mean ± SE (n = 6 per group, except n = 12 for clopidogrel). *P < 0.05 versus the corresponding vehicle (V) (data from Wong et al. [15, 43]). Reproduced from "Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies" published in “Journal of Thrombosis and Haemostasis” (2008), John Wiley and Sons.
Bleeding time studies
The bleeding potential of apixaban was compared with those of rivaroxaban, dabigatran and warfarin in the rabbit cuticle bleeding time model [7, 15]. At the highest effective doses studied (each of which caused ~80% inhibition of thrombus formation), warfarin increased bleeding time almost six-fold, whereas apixaban, rivaroxaban and dabigatran prolonged bleeding time 1.13-, 1.9 and 4.4-fold, respectively (Fig. 3) [7, 15]. As shown in Fig. 3, the antithrombotic efficacy and bleeding profiles of warfarin and dabigatran were less favorable than those of apixaban and rivaroxaban. It should be noted; however, that extrapolation of pre-clinical bleeding time data to humans requires caution. Provoked bleeding measured in anaesthetized healthy animals may not directly translate into spontaneous bleeding observed in the clinical setting, where complications of cardiovascular disease and polypharmacy are often present. Nevertheless, pre-clinical bleeding time studies are still useful for generating hypotheses for clinical investigation, for example by allowing the anti-haemostatic profiles of experimental agents to be ranked and compared with those of established agents such as warfarin. The pre-clinical comparison of these agents’ therapeutic windows, as summarized in Fig. 3, remains a hypothesis, and head-to-head clinical studies are required to validate these results.
Combination therapy
Dual antiplatelet therapy with clopidogrel and aspirin currently represents the standard of care for the reduction of atherothrombotic events in a broad range of patients. To understand the benefit-risk ratio of apixaban therapy in combination with standard antiplatelet therapy, apixaban was evaluated in combination with clinically relevant doses of aspirin and/or clopidogrel for the prevention of arterial thrombosis in rabbit models [44]. These evaluations showed that the triple combination of apixaban, aspirin and clopidogrel resulted in improved antithrombotic activity versus mono-therapies, without excessively increasing bleeding time in rabbits. Such data suggest that intensive antithrombotic therapy with apixaban, aspirin and clopidogrel may be a viable option for enhancing antithrombotic efficacy without unacceptable increases in bleeding.
This hypothesis was tested in a large phase III study, APPRAISE-2, in high-risk patients with recent ACS treated with apixaban or placebo in addition to mono (aspirin) or dual antiplatelet (aspirin plus clopidogrel) therapy. Very recently, the trial was discontinued based on “evidence of a clinically important increase in bleeding among patients randomized to apixaban, and this increase in bleeding was not offset by clinically meaningful reductions in ischemic events” [45]. The investigators of the APPRAISE-2 trial will continue to review the available data to better understand the effects of apixaban in this ACS patient population and will publish the results [45].
As discussed above, the translatability of preclinical bleeding models to safety in clinical settings requires caution. It appears that the preclinical cuticle bleeding effect of apixaban in combination with dual antiplatelet therapy in rabbits does not translate directly into spontaneous bleeding observed in the APPRAISE-2 trial. The underlying causes for this disconnect are not known, but may be related to species differences, bleeding time versus spontaneous bleeding, vascular bed differences, and the fact that unlike animal bleeding models, the APPRAISE-2 patients had the highest tendency to bleed due to advanced age, diabetes, complications of cardiovascular disease, other comorbidities and the additive hazards of combination antiplatelet treatment. Finally, the APPRAISE-2 finding does not mean that apixaban cannot benefit other patient populations, as recent phase III clinical trials of apixaban have demonstrated promising results in patients with venous thromboembolism (ADVANCE 1, 2, 3) and atrial fibrillation (AVERROES) [18, 21, 22, 46].
Ex vivo coagulation markers
The traditional clotting time tests for adjusting anticoagulant doses of heparin (aPTT) and warfarin (PT) are not sensitive for specific, single-target anticoagulants such as the FXa inhibitors. As shown in Fig. 5, apixaban only prolonged ex vivo aPTT and PT modestly, even at the highest dose that produced 80% antithrombotic efficacy in rabbits [7]. As expected from its mechanism of action, apixaban did not prolong thrombin time (TT). Among the clotting time tests, mPT was the most sensitive for apixaban and tracked well with the antithrombotic activity of apixaban. Similar mPT results were also observed with other FXa inhibitors such as rivaroxaban [7]. Data from a phase II study with apixaban show that the anti-FXa assay is more accurate and precise than the mPT test [47]. Indeed, we also observed that the anti-FXa assay tracked well with antithrombotic activity in rabbits with arterial thrombosis [15]. As shown in Fig. 6, apixaban produced a dose-dependent inhibition of FXa and did not inhibit thrombin activity ex vivo [15]. The ex vivo anti-FXa activity of apixaban correlated well with both its antithrombotic activity and plasma concentration (Fig. 6). Thus, the anti-FXa activity assay may be suitable for monitoring the anticoagulant and plasma levels of apixaban if needed in certain situations such as an overdose, acute bleeding or urgent surgery.

Plots of thrombus reduction (bar graph) and ex vivo clotting times (line graph) in apixaban-treated rabbits. Thrombus reduction, measured in the prevention model of venous thrombosis, was expressed as the percentage reduction in thrombus weight after treatment, relative to the mean vehicle thrombus weight (data from Fig. 3). For clarity, only mean data for thrombus reduction and the bolus dose (mg/kg) are shown. Activated partial thromboplastin time (aPTT), prothrombin time (PT), modified prothrombin time (mPT) and thrombin time (TT) were expressed as the treated/control ratio. Data are mean ± SE (n = 6 per group for thrombus reduction and n = 12 per group for clotting times) (data from Wong et al. [7]; reproduced with permission). Reproduced from “Favorable therapeutic index of the direct factor Xa inhibitors, apixaban and rivaroxaban, compared with the thrombin inhibitor dabigatran in rabbits” published in “Journal of Thrombosis and Haemostasis” (2009), John Wiley and Sons

Ex vivo anti-FXa and anti-thrombin effects of apixaban in arterial thrombosis rabbits from Fig. 4 (top) and correlation of ex vivo anti-FXa with antithrombotic effects and plasma concentrations of apixaban in arterial thrombosis rabbits (bottom). *P < 0.05, compared with the vehicle. Mean ± SE and n = 6 per group (data from Wong et al. [15]; reproduced with permission). Reproduced from "Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies" published in “Journal of Thrombosis and Haemostasis” (2008), John Wiley and Sons
Drug metabolism and pharmacokinetics
The metabolism and pharmacokinetics of apixaban have been studied extensively in animals and humans. In these studies, absorption of apixaban after oral administration was rapid, with a time to peak plasma concentration (T max) of 1–2 h. Absolute oral bioavailability of apixaban was good in rats, dogs and humans [48–50]. Following IV administration, apixaban was slowly eliminated in rats, dogs and humans, with an apparent terminal elimination half-life (T 1/2) of 2–11 h, and a total plasma clearance of less than 5% hepatic blood flow. The steady-state volume of distribution for apixaban was low in rats, dogs and humans (approximately 0.31 l/kg) [48–50]. Such steady-state volume of distribution values are indicative of a large portion of the drug remaining in the target compartment (the blood). Apixaban had a higher clearance and a lower bioavailability in rabbits compared with rats, dogs, chimpanzees or humans [48–51]. In humans, apixaban has a low peak-to-trough ratio of approximately 4 or less following oral administration [52, 53]. Serum protein binding did not appear to be concentration dependent in the range of 0.5–5 (μg/ml; protein binding was 92–96% in rats and dogs, respectively, while binding was lower in humans, monkey and rabbit plasma (87, 59 and 63%, respectively) [48]. Table 4 summarizes the pharmacokinetic properties of apixaban in animal species and humans [19, 48–52].
In animals and humans receiving [14C]apixaban, the parent compound was the predominant component in plasma and excreta (urine, bile and feces), although numerous metabolites were detected at relatively low concentrations [51, 53–55]. Metabolic pathways of apixaban in animals and humans are presented in Figs. 7 and 8. In humans, O-demethyl apixaban (M2), O-demethyl apixaban sulfate (M1), 3-hydroxy apixaban (M7) and hydroxylated O-demethyl apixaban (M13) were the most abundant in vivo metabolites. Of these, O-demethyl apixaban sulfate was the predominant circulating human metabolite, with levels of exposure to this metabolite equivalent to approximately 25% of those of apixaban; exposure to other metabolites did not exceed 5% of parent [53]. Overall, approximately 25% of the dose was recovered as metabolites in humans, primarily in the feces. O-Demethyl apixaban followed by O-demethyl apixaban sulfate, 3-hydroxy apixaban and hydroxylated O-demethyl apixaban, were the most abundant metabolites in human excreta. These metabolites were also formed in animal species during non-clinical safety assessments. After administration of [14C]apixaban in mice, rats and dogs, no metabolite exceeded 5% of the total plasma radioactivity at any time point (measured up to 48 h after dosing). While Ο-demethyl apixaban sulfate is the major human circulating metabolite, it does not have meaningful pharmacological activity. In the in vitro enzyme assay, this metabolite did not significantly inhibit purified human FXa at concentrations below 20 μM, and did not inhibit thrombin or trypsin at concentrations up to 30 μM. Furthermore, Ο-demethyl apixaban sulfate does not possess structural alerts and is of no toxicological concern [54].

Biotransformation pathways of apixaban in the mouse, rat, rabbit and dog. Asterisk denotes labeling position of the radioisotope

Biotransformation pathways of apixaban in humans. Asterisk denotes labeling position of the radioisotope
Primary biotransformation reactions of apixaban include O-demethylation and mono-oxidation; in some species, opening of the keto-lactam ring and hydrolysis of the amide moiety are additional minor pathways [55]. Combinations of these reactions were also observed as sulfation of O-demethyl apixaban, sulfation of hydroxylated O-demethyl apixaban and glucuronidation of O-demethyl apixaban (Figs. 7, 8). Apixaban was metabolized very slowly in liver microsomes and hepatocytes, although O-demethyl apixaban was formed in hepatocytes from all species, while O-demethyl apixaban sulfate was detected in rat, monkey and human hepatocytes only. No metabolites were formed by human kidney microsomes or human intestinal S9 fraction. Similarly, no glutathione adduct of apixaban was detected in microsomes or hepatocytes, indicating that the formation of reactive metabolites with apixaban is unlikely. The in vitro metabolism of apixaban was primarily mediated by CYP3A4/5, with relatively minor contributions from CYP1A2 and CYP2J2 towards the formation of O-demethyl apixaban. In addition, low levels of O-demethyl apixaban formation were catalyzed by CYP2C8, CYP2C9 and CYP2C19 [54]. The sulfation of O-demethyl apixaban to form O-demethyl apixaban sulfate, the most abundant circulating metabolite in humans, was primarily catalyzed by the sulfotransferase SULT1A1 [54].
In animals receiving [14C]apixaban, 8.7% (dog) to 47% (rat) of the recovered radioactivity appeared in the urine as apixaban, indicating that renal clearance was a route of apixaban elimination [55]. Biliary clearance was a minor apixaban elimination pathway (<5% of dose in limited collection of bile of rats or humans following oral doses). In bile duct-cannulated rats, 12% of an IV dose was recovered in bile as apixaban (which was also detected in the bile of humans receiving apixaban). Apixaban (at amounts equivalent to 22% of the provided dose) was recovered in the feces after IV administration to bile duct-cannulated rats, suggesting that intestinal secretion of apixaban also occurred. Metabolic clearance was less important than, or of similar magnitude, to non-metabolic clearance (direct excretion/secretion) in rats, dogs and humans. Most of the recovery of metabolites was from the feces. In summary, the elimination of apixaban involves multiple pathways, including hepatic metabolism, renal excretion and intestinal/biliary secretion, each responsible for elimination of approximately one-third of dose.
Apixaban is a substrate for CYP3A4/5, BCRP and P-gp [56]. Co-administration of drugs that modulate CYP3A4/5, P-gp or BCRP activities could therefore potentially affect the disposition of apixaban. Given that apixaban has multiple routes of elimination and an oral bioavailability of approximately 50% [49, 50], any such drug–drug interaction effects are likely to be of relatively low magnitude. This hypothesis is supported by the results of clinical drug–drug interaction studies that show that increases in apixaban exposure are approximately twofold after coadministration with a strong inhibitor of both CYP3A4 and P-gp (i.e. ketoconazole), while an approximately 50% decrease in apixaban exposure is observed after coadministration of apixaban with a strong inducer of both CYP3A4 and P-gp (i.e. rifampin) [50, 57]. The potential of apixaban to inhibit or induce CYP is minimal, suggesting that apixaban is unlikely to affect the metabolism of co-administered medications that are dependent on CYP-mediated clearance.
In summary, orally administered apixaban is well absorbed and bioavailable in humans. The compound has a relatively simple metabolite profile in human plasma, with the only major metabolite an inactive sulfate conjugate. Apixaban is not a significant inhibitor of CYP enzymes or P-gp and so is unlikely to be a significant perpetrator of drug–drug interactions. Apixaban is a substrate for CYP enzymes, BCRP and P-gp, and may show some interaction with drugs that modulate CYP enzymes or these transporters. However, such interactions are unlikely to be of high magnitude since apixaban is eliminated through multiple pathways.
Summary
In summary, apixaban is a novel and potent antithrombotic agent in pre-clinical models. The antithrombotic actions of apixaban are likely related to inhibition of FXa, but not to thrombin inhibition. The high oral bioavailability, low volume of distribution, low plasma clearance and favorable therapeutic index exhibited by apixaban led to its selection for clinical development as an oral anticoagulant. Clinical studies suggest that apixaban may provide consistent anticoagulation and a potentially optimal risk:benefit balance. Phase III studies in patients undergoing total knee replacement have shown that apixaban effectively reduces the risk of venous thromboembolism in this setting, and is associated with lower rates of clinically relevant bleeding than the current standard of care in orthopedic surgery (the low molecular weight heparin enoxaparin) [21, 22]. Other potential indications for apixaban in the prevention and treatment of various life-threatening thromboembolic events are also under investigation in large-scale phase III studies (Table 5).
References
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G et al (2010) American Heart Association Statistics Committee and Stroke Statistics Committee. Executive summary: heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121:948–954
Ruppert A, Lees M, Steinle T (2010) Clinical burden of venous thromboembolism. Curr Med Res Opin 26:2465–2473
Mann KG, Butenas S, Brummel K (2003) The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol 23:17–25
McKenzie CR, Abendschein DR, Eisenberg PR (1996) Sustained inhibition of whole-blood clot procoagulant activity by inhibition of thrombus-associated factor Xa. Arterioscler Thromb Vasc Biol 16:1285–1291
Harker LA, Hanson SR, Kelly AB (1995) Antithrombotic benefits and hemorrhagic risks of direct thrombin antagonists. Thromb Haemost 74:464–472
Wong PC, Crain EJ, Watson CA, Zaspel AM, Wright MR, Lam PY et al (2002) Nonpeptide factor Xa inhibitors III: effects of DPC423, an orally-active pyrazole antithrombotic agent, on arterial thrombosis in rabbits. J Pharmacol Exp Ther 303:993–1000
Wong PC, Crain EJ, Watson CA, Xin B (2009) Favorable therapeutic index of the direct factor Xa inhibitors, apixaban and rivaroxaban, compared with the thrombin inhibitor dabigatran in rabbits. J Thromb Haemost 7:1313–1320
Hauptmann J, Sturzebecher J (1999) Synthetic inhibitors of thrombin and factor Xa: from bench to bedside. Thromb Res 93:203–241
Leadley RJ Jr (2001) Coagulation factor Xa inhibition: biological background and rationale. Curr Top Med Chem 1:151–159
Turpie AG (2004) Fondaparinux: a factor Xa inhibitor for antithrombotic therapy. Expert Opin Pharmacother 5:1373–1384
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G (2008) Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest 133(6 Suppl):160S–198S
FDA News Release, Oct 19, 2010. FDA approves Pradaxa to prevent stroke in people with atrial fibrillation [online]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm230241.htm. Accessed 22 Dec 2010
Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A et al (2007) Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrah ydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem 50:5339–5356
Pinto DJ, Smallheer JM, Cheney DL, Knabb RM, Wexler RR (2010) Factor Xa inhibitors: next-generation antithrombotic agents. J Med Chem 53:6243–6274
Wong PC, Crain EJ, Xin B, Wexler RR, Lam PY, Pinto DJ et al (2008) Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost 6:820–829
APPRAISE Steering Committee and Investigators, Becker RC, Bhatt DL, Cools F, Crea F, Dellborg M et al (2009) Apixaban, an oral, direct, selective factor Xa inhibitor, in combination with antiplatelet therapy after acute coronary syndrome: results of the Apixaban for Prevention of Acute Ischemic and Safety Events (APPRAISE) trial. Circulation 119:2877–2885
Botticelli Investigators Writing Committee, Buller H, Deitchman D, Prins M, Segers A (2008) Efficacy and safety of the oral direct factor Xa inhibitor apixaban for symptomatic deep vein thrombosis. The Botticelli DVT dose-ranging study. J Thromb Haemost 6:1313–1318
Connolly S, Eikelboom J, Flaker G, Kaatz S, Avezum A, Piegas L et al AVERROES: apixaban versus acetylsalicylic acid (ASA) to prevent strokes. ESC 2010 congress reports. European Society of Cardiology web site. http://www.escardio.org/congresses/esc-2010/congress-reports/Pages/708-3-AVERROES.aspx#presenter. Accessed 6 Sept 2010
Frost C, Yu Z, Moore K, Nepal S, Barrett Y, Mosqueda-Garcia R et al (2007) Apixaban, an oral direct factor Xa inhibitor: multiple-dose safety, pharmacokinetics, and pharmacodynamics in healthy subjects. J Thromb Haemost 5(Suppl 2). Abstract PM664
Lassen MR, Davidson BL, Gallus A, Pineo G, Ansell J, Deitchman D (2007) The efficacy and safety of apixaban, an oral, direct factor Xa inhibitor, as thromboprophylaxis in patients following total knee replacement. J Thromb Haemost 5:2368–2375
Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Portman RJ et al (2009) Apixaban or enoxaparin for thromboprophylaxis after knee replacement. N Engl J Med 361:594–604
Lassen MR, Raskob GE, Gallus A, Pineo G, Chen D, Hornick P et al (2010) Apixaban versus enoxaparin for thromboprophylaxis after knee replacement (ADVANCE-2): a randomised double-blind trial. Lancet 375:807–815
Quan ML, Pruitt JR, Ellis CD, Liauw AY, Galemmo RA Jr, Stouten PFW et al (1997) Bisbenzamidine isoxazoline derivatives as factor Xa inhibitors. Bioorg Med Chem Lett 7:2813–2818
Quan ML, Ellis CD, Liauw AY, Alexander RS, Knabb RM, Lam G et al (1999) Design and synthesis of isoxazoline derivatives as factor Xa inhibitors. 2. J Med Chem 42:2760–2773
Quan ML, Liauw AY, Ellis CD, Pruitt JR, Carini DJ, Bostrom LL et al (1999) Design and synthesis of isoxazoline derivatives as factor Xa inhibitors. 1. J Med Chem 42:2752–2759
Wong PC, Quan ML, Crain EJ, Watson CA, Wexler RR, Knabb RM (2000) Nonpeptide factor Xa inhibitors: I. Studies with SF303 and SK549, a new class of potent antithrombotics. J Pharmacol Exp Ther 292:351–357
Pruitt JR, Pinto DJ, Estrella MJ, Bostrom LL, Knabb RM, Wong PC et al (2000) Isoxazolines and isoxazoles as factor Xa inhibitors. Bioorg Med Chem Lett 10:685–689
Pinto DJ, Orwat MJ, Wang S, Fevig JM, Quan ML, Amparo E et al (2001) Discovery of 1-[3-(aminomethyl)phenyl]-N-3-fluoro-2′-(methylsulfonyl)-[1,1′-biphenyl]-4-yl]-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide (DPC423), a highly potent, selective, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem 44:566–578
Wong PC, Pinto DJ, Knabb RM (2002) Nonpeptide factor Xa inhibitors: DPC423, a highly potent and orally bioavailable pyrazole antithrombotic agent. Cardiovasc Drug Rev 20:137–152
Barrett JS, Davidson AF, Jiao QT, Masqueda-Garcia R, Kornhauser DM, Gangrade NK et al (2001) The effect of food, formulation, and dosing duration on the pharmacokinetics of DPC-423, a potent factor Xa inhibitor (abstract). J Clin Pharmacol 41:1023
Quan ML, Lam PY, Han Q, Pinto DJ, He MY, Li R et al (2005) Discovery of 1-(3′-aminobenzisoxazol-5′-yl)-3-trifluoromethyl-N-[2-fluoro-4-[(2′-dimethylaminomethyl)imidazol-1-yl]phenyl]-1H-pyrazole-5-carboxyamide hydrochloride (razaxaban), a highly potent, selective, and orally bioavailable factor Xa inhibitor. J Med Chem 48:1729–1744
Wong PC, Crain EJ, Watson CA, Wexler RR, Lam PY, Quan ML et al (2007) Razaxaban, a direct factor Xa inhibitor, in combination with aspirin and/or clopidogrel improves low-dose antithrombotic activity without enhancing bleeding liability in rabbits. J Thromb Thrombolysis 24:43–51
Lassen MR, Davidson BL, Gallus A, Pineo G, Ansell J, Deitchman D (2003) A phase II randomized, double-blind, five-arm, parallel-group, dose-response study of a new oral directly-acting factor Xa inhibitor, razaxaban, for the prevention of deep vein thrombosis in knee replacement surgery—on behalf of the razaxaban investigators. Blood (ASH Annual Meeting Abstracts) 102:111. Abstract 41
Pinto DJ, Orwat MJ, Quan ML, Han Q, Galemmo RA Jr, Amparo E et al (2006) 1-[3-Aminobenzisoxazol-5′-yl]-3-trifluoromethyl-6-[2′-(3-(R)-hydroxy-N-pyr rolidinyl)methyl-[1,1′]-biphen-4-yl]-1,4,5,6-tetrahydropyrazolo-[3,4-c]-py ridin-7-one (BMS-740808) a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. Bioorg Med Chem Lett 16:4141–4147
Luettgen JM, Knabb RM, He K, Pinto DJ, Rendina AR (2011) Apixaban inhibition of factor Xa: Microscopic rate constants and inhibition mechanism in purified protein systems and in human plasma. J Enzyme Inhib Med Chem. Posted online on 20 Dec 2010. doi:10.3109/14756366.2010.535793
Krishnaswamy S (2005) Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost 3:54–67
Jiang X, Crain EJ, Luettgen JM, Schumacher WA, Wong PC (2009) Apixaban, an oral direct factor Xa inhibitor, inhibits human clot-bound factor Xa activity in vitro. Thromb Haemost 101:780–782
Hara T, Yokoyama A, Morishima Y, Kunitada S (1995) Species differences in anticoagulant and anti-Xa activity of DX-9065a, a highly selective factor Xa inhibitor. Thromb Res 80:99–104
Perzborn E, Strassburger J, Wilmen A, Pohlmann J, Roehrig S, Schlemmer KH et al (2005) In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct factor Xa inhibitor. J Thromb Haemost 3:514–521
Luettgen J, Wang Z, Seiffert D, Rendina A, Knabb R, Ogletree M (2007) Inhibition of measured thrombin generation in human plasma by apixaban: a predictive mathematical model based on experimentally determined rate constants. J Thromb Haemost 5(Suppl 2). Abstract PT633
Wong PC, Jiang X (2010) Apixaban, a direct factor Xa inhibitor, inhibits tissue-factor induced human platelet aggregation in vitro: comparison with direct inhibitors of factor VIIa, XIa and thrombin. Thromb Haemost 104:302–310
Schumacher WA, Bostwick JS, Stewart AB, Steinbacher TE, Xin B, Wong PC (2010) Effect of the direct factor Xa inhibitor apixaban in rat models of thrombosis and hemostasis. J Cardiovasc Pharmacol 55:609–616
Wong PC, Crain EJ, Watson CA, Hua J, Schumacher WA, Rehfuss R (2009) Clopidogrel versus prasugrel in rabbits. Effects on thrombosis, haemostasis, platelet function and response variability. Thromb Haemost 101:108–115
Wong PC, Watson CA, Crain EJ (2008) Arterial antithrombotic and bleeding time effects of apixaban, a direct factor Xa inhibitor, in combination with antiplatelet therapy in rabbits. J Thromb Haemost 6:1736–1741
Bristol Myers Squibb Press Releases, Nov 18, 2010. APPRAISE-2 study with investigational compound apixaban in acute coronary syndrome discontinued [online]. http://www.bms.com/news/press_releases/pages/default.aspx?RSSLink=http://www.businesswire.com/news/bms/20101118007161/en&t=634266370298980311. Accessed 29 Nov 2010
Lassen MR, Gallus A, Raskob GE, Pineo G, Chen D, Ramirez LM, ADVANCE-3 Investigators (2010) Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N Engl J Med 363(26):2487–2498
Barrett YC, Wang Z, Frost C, Shenker A (2010) Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 104:1263–1271
He K, He B, Grace JE, Xin B, Zhang D, Pinto D et al (2006) Preclinical pharmacokinetics and metabolism of apixaban, a potent and selective factor Xa inhibitor. Blood (ASH Annual Meeting Abstracts) 108. Abstract 910
Frost C, Yu Z, Nepal S, Bragat A, Moore K, Shenker A et al (2008) Apixaban, a direct factor Xa inhibitor: single-dose pharmacokinetics and pharmacodynamics of an intravenous formulation. J Clin Pharmacol 48:1132. Abstract 142
Vakkalagadda B, Frost C, Wang J, Nepal S, Schuster A, Zhang D et al (2009) Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. J Clin Pharamcol 49:1124. Abstract 143
Zhang D, He K, Raghavan N, Wang L, Crain EJ, He B et al (2010) Metabolism, pharmacokinetics and pharmacodynamics of the factor Xa inhibitor apixaban in rabbits. J Thromb Thrombolysis 29:70–80
Frost C, Yu Z, Nepal S, Mosqueda-Garcia R, Shenker A (2007) Apixaban, an oral direct, factor Xa inhibitor: single-dose safety, pharmacokinetics and pharmacodynamics in healthy subjects. J Thromb Haemost 5(Suppl 2). Abstract P-M-665
Raghavan N, Frost CE, Yu Z, He K, Zhang H, Humphreys WG et al (2009) Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos 37(1):74–81
Wang L, Raghavan N, He K, Luettgen JM, Humphreys WG, Knabb RM et al (2009) Sulfation of o-demethyl apixaban: enzyme identification and species comparison. Drug Metab Dispos 37:802–808
Zhang D, He K, Raghavan N, Wang L, Mitroka J, Maxwell BD et al (2009) Comparative metabolism of 14C-labeled apixaban in mice, rats, rabbits, dogs, and humans. Drug Metab Dispos 37:1738–1748
Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE et al (2010) In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos 38:448–458
Frost C, Wang J, Nepal S, Schuster A, Zhang D, Yu Z et al (2009) Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. J Clin Pharmacol 49:1123. Abstract 139
Acknowledgments
The authors wrote the first draft of the paper and reviewed and modified all subsequent drafts. Rick Flemming, at PPSI, a PAREXEL company, provided professional writing and editorial assistance, which was funded by Bristol-Myers Squibb and Pfizer. The authors thank Charles Frost, W Griffith Humphreys, Joseph Luettgen and William Schumacher for critical review of the manuscript. The authors thank Kan He, Robert Knabb, Patrick Lam, Joseph Luettgen, Michael Orwat, Mimi Quan, Lucius Rossano and Ruth Wexler for their contributions to the preclinical discovery of apxiaban.
Conflict of interest
The authors are employees of Bristol-Myers Squibb Company. This research and review were funded by Bristol-Myers Squibb and Pfizer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wong, P.C., Pinto, D.J.P. & Zhang, D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis 31, 478–492 (2011). https://doi.org/10.1007/s11239-011-0551-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-011-0551-3