
Abstract
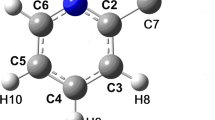
In this study, the molecular structure of p-diisocyanobenzene has been determined by gas-phase electron diffraction and quantum chemical calculations. The electron diffraction intensities from a previous study by Colapietro et al. (J Mol Struct 125:19–32, 1984) have been reanalyzed using geometrical constraints and initial values of vibrational amplitudes from computations. The equilibrium structure of the molecule has D 2h symmetry, whereas the average geometry in the gaseous phase is best described by a non-planar model of C 2v symmetry. The lowering of symmetry is due to large-amplitude motion of the substituents out of the plane of the benzene ring. The non-planar model has an internal ring angle at the ipso position, ∠aC2–C1–C6 = 120.6 ± 0.2°, about 1° smaller than that from the previous study, but consistent with the quantum chemical calculations. The mean length of the ring C–C bonds and the length of the triple bond are accurately determined as 〈r g(C–C)〉 = 1.398 ± 0.003 Å and r g(N≡C) = 1.177 ± 0.002 Å, respectively. Comparison with the gaseous isoelectronic molecules p-diethynylbenzene and p-dicyanobenzene shows that the differences in the mean lengths of the ring C–C bonds and in the lengths of the triple bonds determined by electron diffraction are equal or closely similar to the corresponding differences from quantum chemical calculations. The present experimental value of the ipso angle in free p-diisocyanobenzene is slightly, but significantly smaller than that obtained by X-ray crystallography. The difference is confirmed by computational modeling of the crystal structure and appears to be due to –N≡C···H–C intermolecular interactions in the crystal.






Similar content being viewed by others

References
Domenicano A, Murray-Rust P, Vaciago A (1983) Acta Crystallogr B 39:457–468
Campanelli AR, Domenicano A, Ramondo F (2003) J Phys Chem A 107:6429–6440
Campanelli AR, Domenicano A, Ramondo F, Hargittai I (2004) J Phys Chem A 108:4940–4948
Campanelli AR, Domenicano A, Macchiagodena M, Ramondo F (2011) Struct Chem 22:1131–1141
Domenicano A, Arcadi A, Ramondo F, Campanelli AR, Portalone G, Schultz G, Hargittai I (1996) J Phys Chem 100:14625–14629
Campanelli AR, Arcadi A, Domenicano A, Ramondo F, Hargittai I (2006) J Phys Chem A 110:2045–2052
Campanelli AR, Domenicano A, Ramondo F, Hargittai I (2008) J Phys Chem A 112:10998–11008
Colapietro M, Domenicano A, Portalone G, Torrini I, Hargittai I, Schultz G (1984) J Mol Struct 125:19–32
Poirier R, Kari R, Csizmadia IG (1985) Handbook of Gaussian basis sets: a compendium for ab initio molecular orbital calculations. Elsevier, Amsterdam
Dunning TH Jr (1989) J Chem Phys 90:1007–1023
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C.02. Gaussian, Inc., Wallingford
Moran D, Simmonett AC, Leach FE III, Allen WD, Schleyer PvR, Schaefer HF III (2006) J Am Chem Soc 128:9342–9343
Hedberg L, Mills IM (2000) J Mol Spectrosc 203:82–95
Hedberg L, Mills IM (1993) J Mol Spectrosc 160:117–142
Hargittai I, Hernádi J, Kolonits M (1972) Prib Tekh Eksp 239–240
Tremmel J, Kolonits M, Hargittai I (1977) J Phys E 10:664
Hargittai I, Tremmel J, Kolonits M (1980) Hung Sci Instrum 50:31–42
Domenicano A, Schultz G, Kolonits M, Hargittai I (1979) J Mol Struct 53:197–209
Colapietro M, Domenicano A, Portalone G, Schultz G, Hargittai I (1987) J Phys Chem 91:1728–1737
Andersen B, Seip HM, Strand TG, Stølevik R (1969) Acta Chem Scand 23:3224–3234
Portalone G, Domenicano A, Schultz G, Hargittai I (1989) J Mol Struct (Theochem) 186:185–196
Gauss J, Stanton JF (2000) J Phys Chem A 104:2865–2868
Plíva J, Johns JWC, Goodman L (1991) J Mol Spectrosc 148:427–435
Maslen PE, Handy NC, Amos RD, Jayatilaka D (1992) J Chem Phys 97:4233–4254
Colapietro M, Domenicano A, Portalone G, Schultz G, Hargittai I (1984) J Mol Struct 112:141–157
Campanelli AR, Domenicano A, Ramondo F (2006) J Phys Chem A 110:10122–10129
Domenicano A (1992) In: Domenicano A, Hargittai I (eds) Accurate molecular structures: their determination and importance, Chap. 18. Oxford University Press, Oxford, pp 437–468
Gillespie RJ, Hargittai I (1991) The VSEPR model of molecular geometry. Allyn and Bacon, Boston; (2012) Dover, Mineola, NY
Acknowledgments
This study was supported by the CASPUR Supercomputing Center, Rome, with Standard HPC Grant 2011 (“A combined X-ray absorption spectroscopy, molecular dynamics simulations, and quantum mechanics calculation procedure for the structural characterization of ill-defined systems”).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Campanelli, A.R., Domenicano, A., Ramondo, F. et al. Molecular structure of p-diisocyanobenzene from gas-phase electron diffraction and theoretical calculations and effects of intermolecular interactions in the crystal on the benzene ring geometry. Struct Chem 23, 287–295 (2012). https://doi.org/10.1007/s11224-011-9889-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-011-9889-6